Standard and New Cancer Drugs
New Cancer Drugs/Immunotherapy/Targeted Therapy
Standard Cancer Drugs/Chemotherapy
Immune checkpoint inhibitor
A type of drug that blocks certain proteins made by some types of immune system cells, such as T cells, and some cancer cells. These proteins help keep immune responses in check and can keep T cells from killing cancer cells. When these proteins are blocked, the “brakes” on the immune system are released and T cells are able to kill cancer cells better. Examples of checkpoint proteins found on T cells or cancer cells include PD-1/PD-L1 and CTLA-4. Some immune checkpoint inhibitors are used to treat cancer.

Immune checkpoint inhibitor
Checkpoint proteins, such as PD-L1 on tumor cells and PD-1 on T cells, help keep immune responses in check. The binding of PD-L1 to PD-1 keeps T cells from killing tumor cells in the body (left panel). Blocking the binding of PD-L1 to PD-1 with an immune checkpoint inhibitor (anti-PD-L1 or anti-PD-1) allows the T cells to kill tumor cells (right panel).

Functions of CD28, B7-1, B7-2, and CTLA-4 Molecules (CTLA-4 is an immune checkpoint protein)
Resting T cells express CD28, but resting antigen-presenting cells (APCs) do not express B7 molecules. Within six hours after activation, B7- 2 is expressed by antigen-presenting cells and is available to bind to CD28, ransmitting a costimulatory signal to the T cell. By 48 to 72 hours after activation, antigen-presenting cells also express B7-1, whereas T cells express the CTLA-4 inhibitory receptor. Both B7-1 and B7-2 can bind to either CD28 or CTLA-4, providing continued costimulation or a new inhibitory signal, respectively. Because CTLA-4 binds B7 molecules with a higher affinity than does CD28, its inhibitory interaction eventually predominates, leading to the termination of the immune response. The fusion protein CTLA-4ÐIg can compete with CD28 and CTLA-4 for B7 binding, thus preventing costimulatory interactions.
Immune checkpoint inhibitors to treat cancer
Drugs that target PD-1 or PD-L1
PD-1 is a checkpoint protein on immune cells called T cells. It normally acts as a type of “off switch” that helps keep the T cells from attacking other cells in the body. It does this when it attaches to PD-L1, a protein on some normal (and cancer) cells. When PD-1 binds to PD-L1, it basically tells the T cell to leave the other cell alone. Some cancer cells have large amounts of PD-L1, which helps them evade immune attack.
Monoclonal antibodies that target either PD-1 or PD-L1 can block this binding and boost the immune response against cancer cells. These drugs have shown a great deal of promise in treating certain cancers.
# PD-1 inhibitors: Examples of drugs that target PD-1 include:
■ Pembrolizumab (Keytruda)
■ Nivolumab (Opdivo)
These drugs have been shown to be helpful in treating several types of cancer, including melanoma of the skin, non-small cell lung cancer, kidney cancer, bladder cancer, head and neck cancers, and Hodgkin lymphoma. They are also being studied for use against many other types of cancer.
# PD-L1 inhibitors: Examples of drugs that target PD-L1 include:
■ Atezolizumab (Tecentriq)
■ Avelumab (Bavencio)
■ Durvalumab (Imfinzi)
These drugs have also been shown to be helpful in treating different types of cancer, including bladder cancer, non-small cell lung cancer, and Merkel cell skin cancer (Merkel cell carcinoma). They are also being studied for use against other types of cancer.
One concern with all of these drugs is that they can allow the immune system to attack some normal organs in the body, which can lead to serious side effects in some people. Common side effects of these drugs can include fatigue, cough, nausea, loss of appetite, skin rash, and itching. Less often they can cause more serious problems in the lungs, intestines, liver, kidneys, hormone-making glands, or other organs.
Many other drugs that target either PD-1 or PD-L1 are now being tested in clinical trials as well, both alone and combined with other drugs (see What’s new in cancer immunotherapy research?).
Drugs that target CTLA-4
CTLA-4 is another protein on some T cells that acts as a type of “off switch” to keep the immune system in check.
■ Ipilimumab (Yervoy) is a monoclonal antibody that attaches to CTLA-4 and stops it from working. This can boost the body’s immune response against cancer cells.
This drug is used to treat melanoma of the skin. It is also being studied for use against other cancers.
Because ipilimumab affects the immune system, it can sometimes cause serious or even life-threatening side effects. In fact, compared to drugs that target PD-1 or PD-L1, serious side effects seem to be more likely with ipilimumab.
| Name | Target | Approved | Company |
| Ipilimumab (Yervoy) | CTLA-4 | 2011 | Bristol-MS |
| Nivolumab (Opdivo) | PD-1 | 2014 | Bristol-MS /MDX |
| Pembrolizumab (Keytruda) | PD-1 | 2014 | Merck |
| Atezolizumab (Tecentriq) | PD-L1 | 2016 | Roche/ Genentech |
| Avelumab (Bavencio) | PD-L1 | 2017 | Merck/Pfizer/Eli Lilly |
| Durvalumab (Imfinzi) | PD-L1 | 2017 | Medimmune/AstraZeneca |
The first checkpoint antibody approved by the FDA was ipilimumab, approved in 2011 for treatment of melanoma. Clinical trials have also shown some benefits of anti-CTLA-4 therapy on lung cancer or pancreatic cancer, specifically in combination with other drugs
STORAGE
- Diluted product should be used within 6 hours if stored at room temperature or within 24 hours if stored at 36 to 46 degrees F
- Discard product if it contains particulate matter, is cloudy, or discolored
- Discard unused portion. Do not store for later use.
- Do not freeze
- Protect from light
- Refrigerated product should reach room temperature before administration
- Store in original carton in refrigerator (35 to 46 degrees F) until time of use
Mix diluted solution by gentle inversion. Do not shake.
- Diluted product is stable and sterile for 24 hours when stored refrigerated or at room temperature
- Discard product if it contains particulate matter, is cloudy, or discolored
- Protect from light
Do not administer other drugs through the same infusion line.
Administer Antineoplastic Monoclonal Antibodies prior to chemotherapy when given on the same day.
† Indicates off-label use
Ipilimumab (Yervoy)
Monoclonal antibody that binds to the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and blocks its interaction with its ligands CD80/CD86.
Indicated for malignant melanoma.
May cause fatal immune-mediated adverse reactions such as enterocolitis, hepatitis, dermatitis, neuropathy, and endocrinopathy.
Immune-mediated reactions
Ipilimumab administration can result in severe and fatal immune-mediated reactions. Reactions usually manifest during treatment although a minority have been reported weeks to months after treatment discontinuation. While any organ system can be affected, the most common severe reactions are enterocolitis, hepatitis, dermatitis (including toxic epidermal necrolysis), neuropathy, and endocrinopathy.
Prior to treatment initiation and before each dose is administered, asses for signs and symptoms of enterocolitis, dermatitis, neuropathy, and endocrinopathy; also, evaluate clinical blood chemistries including liver function tests, adrenocorticotropic hormone (ACTH) concentrations, and thyroid function tests.
Permanently discontinue ipilimumab and initiate systemic high-dose corticosteroid therapy for severe immune-mediated reactions.
IgG1
Yervoy Intravenous Inj Sol 5mg/ml is supplied in 2 quantities 200mg/40ml and 50mg/10ml, stored refrigerated at 2-8o C.
Visually inspect parenteral products for particulate matter and discoloration prior to administration whenever solution and container permit. The ipilimumab solution may have a pale yellow color and may have translucent-to-white, amorphous particles. Discard the ipilimumab vial if the solution is cloudy, if there is pronounced discoloration, or if there is foreign particulate matter.
Intravenous Administration
Preparation:
Allow the ipilimumab vials to stand at room temperature for approximately 5 minutes before infusion preparation.
Withdraw the required volume of ipilimumab and transfer into an intravenous bag. Discard partially used vials or empty vials of ipilimumab.
Dilute with 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP to prepare a diluted solution with a final ipilimumab concentration ranging from 1 to 2 mg/mL. Mix diluted solution by gentle inversion. Do not shake. Do not mix ipilimumab with other medicinal products.
Storage: Once diluted, store for no more than 24 hours under refrigeration (2 to 8 degrees C or 36 to 46 degrees F) or at room temperature (20 to 25 degrees C or 68 to 77 degrees F).
Intravenous infusion:
Administer the diluted infusion over 90 minutes through an intravenous line containing a sterile, non-pyrogenic, low-protein-binding, in-line filter. Do not administer as an infusion with other medicinal products.
After each infusion, flush the intravenous line with 0.9% Sodium Chloride Injection, USP or 0.5% Dextrose Injection, USP.
■ For the treatment of unresectable or metastatic melanoma, as a single-agent.
Adults, Adolescents, and Children 12 years of age.
3 mg/kg IV over 90 minutes repeated every 3 weeks for a total of 4 doses.
Doses may be delayed for toxicity; however, all treatment must be administered within 16 weeks of the first dose.
■ For the adjuvant treatment of cutaneous melanoma with pathologic involvement of regional lymph nodes of more than 1 mm, in patients who have undergone complete resection, including total lymphadenectomy.
10 mg/kg IV over 90 minutes every 3 weeks for 4 doses, followed by 10 mg/kg IV every 12 weeks until disease recurrence or unacceptable toxicity, for up to 3 years.
In the event of toxicity, doses may be omitted but not delayed.
† For the first-line treatment of unresectable or metastatic melanoma, in combination with dacarbazine.
10 mg/kg IV plus dacarbazine 850 mg/m2 IV repeated every 3 weeks (at weeks 1, 4, 7, and 10) for 4 doses followed by dacarbazine 850 mg/m2 IV every 3 weeks through week 22 (if no progressive disease) as induction therapy.
At week 24, patients with stable disease or an objective response received maintenance therapy with ipilimumab 10 mg/kg IV every 12 weeks until progressive disease.
(Note: Due to higher risk of serious immune-mediated toxicities, this regimen is not used in clinical practice.)
† For the treatment of unresectable or metastatic melanoma, in combination with nivolumab.
■ NOTE: Nivolumab is FDA approved in combination with ipilimumab for the treatment of unresectable or metastatic malignant melanoma.
3 mg/kg IV over 90 minutes plus nivolumab 1 mg/kg IV over 60 minutes repeated every 3 weeks for 4 doses followed by nivolumab 240 mg IV over 60 minutes repeated every 2 weeks until disease progression or unacceptable toxicity.
Administer ipilumumab after nivolumab. Both agents may need to be temporarily withheld or permanently discontinued in patients who develop immune-related reactions. Interrupt or slow the rate of the nivolumab infusion in patients who develop mild or moderate infusion reactions; discontinue therapy for severe or life-threatening infusion-related reactions.
† For the treatment of unresectable or metastatic melanoma, following no more than 1 prior therapy, in combination with sargramostim (GM-CSF).
10 mg/kg (actual body weight) IV on day 1 repeated every 3 weeks for 4 cycles in combination with sargramostim 250 micrograms (mcg) subcutaneously on days 1 to 14 repeated every 3 weeks for 4 cycles as induction therapy.
In patients with stable disease or better, maintenance therapy consisted of ipilimumab 10 mg/kg (actual body weight) IV on day 1 repeated every 12 weeks (starting on cycle 8) in combination with sargramostim 250 mcg subcutaneously on days 1 to 14 repeated every 3 weeks (starting on cycle 5). At a median follow-up of 13.3 months, median OS was significantly improved with combination therapy of ipilimumab plus GM-CSF compared to single-agent ipilimumab (17.5 months vs. 12.7 months; p = 0.01).
† For the treatment of unresectable advanced melanoma in patients eligible for local treatment of cutaneous, subcutaneous, and nodal lesions, in combination with talimogene laherparepvec†.
Dosage not established. Although overall response rate was improved with talimogene laherparepvec plus ipilimumab therapy compared with ipilimumab alone in a randomized clinical trial; there is not sufficient evidence to support the use of this drug combination for this indication.
Ipilimumab 3 mg/kg IV every 3 weeks beginning on day 1 of week 6 for up to 4 infusions in combination with talimogene laherparepvec (given as a dose volume up to 4 mL at a concentration of 1 million plaque-forming units (PFU)/mL intralesionally on day 1 of week 1 followed by up to 4 mL at a concentration of 100 million PFU/mL on day 1 of week 4 and then every 2 weeks until complete response, all injectable tumors have disappeared, confirmed disease progression per modified immune-related response criteria, or intolerance) was evaluated in a multinational, randomized, phase 2 trial (n = 198).[63055]
■ For the first-line treatment of intermediate or poor risk advanced renal cell cancer (RCC), in combination with nivolumab.
Ipilimumab 1 mg/kg IV over 30 minutes on day 1, preceded by nivolumab 3 mg/kg IV over 30 minutes, every 3 weeks for 4 doses.
After completion of 4 doses of nivolumab plus ipilimumab, continue nivolumab as monotherapy until disease progression or unacceptable toxicity.
Therapy may need to be temporarily withheld or permanently discontinued in patients who develop immune-related reactions. Interrupt or slow the rate of the infusion in patients who develop mild or moderate infusion reactions; discontinue therapy for severe or life-threatening infusion-related reactions.
In a randomized, open-label study, treatment with nivolumab plus ipilimumab significantly improved median overall survival (not estimated vs. 25.9 months) and objective response rate (41.6% vs. 26.5%) compared with sunitinib in patients with intermediate/poor risk patients with previously untreated RCC; a complete response was achieved in 9.4% of patients in the nivolumab plus ipilimumab arm compared with 1.2% of those who received sunitinib. Median progression-free survival was 11.6 months compared with 8.4 months, respectively.
In a separate analysis, the combination of nivolumab plus ipilimumab in patients with favorable risk disease did not significantly improve overall survival; efficacy in this population has not been established.
■ For the treatment of microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan, in combination with nivolumab.
Ipilimumab 1 mg/kg IV over 30 minutes following administration of nivolumab 3 mg/kg IV over 30 minutes, on day 1 every 3 weeks for 4 cycles. After completion of 4 cycles of combination therapy, continue nivolumab 240 mg IV over 30 minutes as a single agent every 2 weeks until disease progression or unacceptable toxicity.
Therapy may need to be temporarily withheld or permanently discontinued ........
In a multicenter, non-randomized, multiple parallel-cohort, open-label study, treatment with nivolumab plus ipilimumab resulted in an overall response rate by independent radiographic review of 46% in patients with locally determined dMMR or MSI-H metastatic colorectal cancer who had received prior treatment with a fluoropyrimidine, oxaliplatin, and irinotecan (n = 82), with 43% partial responses. The duration of response was 6 months or longer in 89% of these patients, and was 12 months or longer in 21% of patients.
Children and Adolescents 12 to 17 years
Ipilimumab 1 mg/kg IV over 30 minutes following administration of nivolumab 3 mg/kg IV over 30 minutes, on day 1 every 3 weeks for 4 cycles. After completion of 4 cycles of combination therapy, continue nivolumab 240 mg IV over 30 minutes as a single agent every 2 weeks until disease progression or unacceptable toxicity.Ipilimumab
Therapy may need to be temporarily withheld or permanently discontinued in patients who develop immune-related reactions. Interrupt or slow the rate of the infusion in patients who develop mild or moderate infusion reactions; discontinue therapy for severe or life-threatening infusion-related reactions. Efficacy for pediatric patients has been extrapolated from results from clinical trials in adult MSI-H or dMMR metastatic colorectal cancer patients with additional population pharmacokinetic data. In a multicenter, non-randomized, multiple parallel-cohort, open-label study, treatment with nivolumab plus ipilimumab resulted in an overall response rate by independent radiographic review of 46% in patients with locally determined dMMR or MSI-H metastatic colorectal cancer who had received prior treatment with a fluoropyrimidine, oxaliplatin, and irinotecan (n = 82), with 43% partial responses. The duration of response was 6 months or longer in 89% of these patients, and was 12 months or longer in 21% of patients.
Nivolumab (Opdivo)
Programmed death receptor-1 (PD-1) blocking human monoclonal antibody.
Used for certain types of melanoma, non-small cell lung cancer, head and neck cancer, renal cell carcinoma, Hodgkin lymphoma, urothelial carcinoma, colorectal cancer, and hepatocellular carcinoma.
Serious immune-mediated adverse reactions (e.g., pneumonitis, colitis, hepatitis, nephritis/renal dysfunction, hypo-/hyperthyroidism) have been reported.
Nivolumab is a fully human IgG4 monoclonal antibody.
Nivolumab/Opdivo Intravenous Inj Sol: 10mg/ml, stored refrigerated (between 36 and 46 degrees F).
Preparation:
Withdraw the required volume of drug and transfer into an intravenous container.
Dilute nivolumab with either 0.9% sodium chloride injection or 5% dextrose injection, to prepare an infusion with a final concentration ranging from 1 to 10 mg/mL. The total volume must not exceed 160 mL.
Mix the diluted solution by gentle inversion. Do not shake.
Discard partially used or empty vials of nivolumab.
Storage: After preparation, store either at room temperature for no more than 4 hours (this includes storage time in the IV container and time for administration of the infusion) or under refrigeration (2 to 8 degrees C or 36 to 46 degrees F) for no more than 24 hours.
STANDARD TREATMENT REGIMEN:
240 mg IV over 30 minutes every 2 weeks OR 480 mg IV over 30 minutes every 4 weeks, until disease progression or unacceptable toxicity.
Therapy may need to be temporarily withheld or permanently discontinued in patients who develop immune-related reactions.
Interrupt or slow the rate of the infusion in patients who develop mild or moderate infusion reactions; discontinue therapy for severe or life-threatening infusion-related reactions.
For the treatment of malignant melanoma.
■ For the treatment of BRAF V600 mutation-positive unresectable or metastatic melanoma
Stage IIIc or IV melanoma, phase III
Nivolumab compared with investigator's choice chemotherapy (ICC)
At a follow-up of approximately 2 years, the median overall survival (15.7 months vs. 14.4 months), Objective response rate (27% vs 10%)
the median durations of response were 32 months and 13 months, respectively.
ICC chemotherapy consisted of dacarbazine 1,000 mg/m2 IV every 3 weeks or carboplatin (AUC 6) plus paclitaxel 175 mg/m2 every 3 weeks
■ For the treatment of BRAF V600 wild-type unresectable or metastatic melanoma as single-agent therapy.
■ For the treatment of unresectable or metastatic melanoma, in combination with ipilimumab.
Nivolumab 1 mg/kg IV over 30 minutes followed by ipilimumab 3 mg/kg IV over 90 minutes repeated every 3 weeks for 4 doses,
followed by standard nivolumab regimen.
■ For the adjuvant treatment of melanoma in patients with lymph node involvement or metastatic disease who have undergone complete resection.
Standard regimen for up to 1 year.
■ For the treatment of metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy, and after progression on EGFR- or ALK-targeted therapy if applicable.
Standard regimen compared with docetaxel.
A statistically significant improvement in overall survival (OS) and progression-free survival (PFS) compared with docetaxel.
PD-L1 expression did not correlate with significantly improved OS.
OS was also significantly improved in patients with platinum-resistant metastatic non-squamous NSCLC.
In a separate randomized, open-label study; the objective response rate was 19% vs. 12%, with a median duration of response of 17 months vs. 6 months, respectively. PFS was not improved in the nivolumab arm. In this study, PD-L1 expression correlated with improved outcomes in both OS and PFS.
In a pooled analysis providing a minimum of 2 years follow-up for these studies, OS with nivolumab versus docetaxel was 23% vs. 8% in patients with squamous NSCLC, and 29% vs. 16% in non-squamous NSCLC. Ongoing responses after 2 years of follow-up were evident in 37% of nivolumab-treated patients with squamous NSCLC and 34% of patients with non-squamous NSCLC; no patients who received docetaxel had an ongoing response.
■ For the treatment of metastatic small cell lung cancer (SCLC), after progression on platinum-based chemotherapy and at least one other line of therapy.
Nivolumab 240 mg IV over 30 minutes on day 1, then every 2 weeks until disease progression or unacceptable toxicity.
A multicenter, open-label, open-cohort ongoing clinical trial evaluated patients with advanced or metastatic solid tumors treated with nivolumab as a single agent or in combination with ipilimumab. In this trial, patients with metastatic SCLC and progression after platinum-based chemotherapy and at least one other line of therapy who received with nivolumab (n = 109) had an overall response rate of 12% (complete response, 0.9%; partial response, 11%), regardless of PD-L1 status. The duration of response was 6 months or longer in 77% of patients, 12 months or longer in 62% of patients, and 18 months or longer in 38% of patients.[58668]
■ For the treatment of advanced renal cell cancer (RCC) who have received prior anti-angiogenic therapy.
Standard regimen compared with everolimus 10 mg once daily.
The primary outcome of overall survival was significantly improved in patients who received nivolumab compared with everolimus (25 vs. 19.6 months), regardless of PD-L1 expression level. The confirmed objective response rate (ORR) was 21.5% in nivolumab-treated patients with a median time to onset of 3 months, compared to 3.9% ORR in those who received everolimus and a time to onset of 3.7 months. Responses lasted for a median duration of 23 months and 13.7 months, respectively.
■ For the first-line treatment of intermediate or poor risk advanced renal cell cancer (RCC), in combination with ipilimumab.
See ipilimumab
■ For the treatment of classical Hodgkin lymphoma that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin.
Standard regimen
Phase I trial (n = 23) in a cohort of patients with relapsed or refractory classical Hodgkin lymphoma . ORR was 87%. Median therapy duration of 36 weeks, (range 13 to 77 weeks). The complete response (CR) rate was 17%.
In 15 patients that had previously received an autologous HSCT and post-transplant brentuximab vedotin, the ORR was 87% and the CR rate was 7%. At a median follow-up time of 40 weeks (range, 0 to 75 weeks), the median overall survival (OS) time had not been reached and the 24-week progression-free survival (PFS) rate was 86%. In this study, 78% of patients had previously received brentuximab vedotin therapy, 78% of patients had undergone a prior autologous HSCT, and 65% of patients had received 4 or more prior therapies.
In a multinational, multicohort, phase II trial, the ORR (primary endpoint assessed by an independent radiological review committee) was 66.3% in 80 patients with classical HL who had failed to respond to autologous SCT and had either relapsed after or failed to respond to brentuximab vedotin; the CR rate was 9% in these patients. The median response duration was 7.8 months. All patients (median age, 37 years) had previously received brentuximab vedotin; patients had received a median of 4 prior therapies. At a median follow-up of 8.9 months, the 6-month PFS and OS rates were 76.9% and 98.7%, respectively. At 12 months, the median PFS time was 10 months.
■ For the treatment of classical Hodgkin lymphoma that has relapsed or progressed after 3 or more lines of systemic therapy that includes an autologous hematopoietic stem cell transplantation (HSCT).
Standard regimen
In a pooled analysis from 2 clinical studies (n = 258), the objective response rate was 69% in patients who had relapsed or progressive classical Hodgkin lymphoma following an autologous HSCT; the complete remission rate was 14%. In this analysis, patients had received a median of 4 prior systemic regimens (range, 2 to 15 regimens) and 76% of patients had received prior brentuximab vedotin.
■ For the treatment of recurrent or metastatic head and neck cancer (squamous cell) with disease progression on or after platinum-containing chemotherapy.
Standard regimen
In a multicenter, randomized, open-label clinical trial, nivolumab significantly improved overall survival compared with investigator’s choice of weekly monotherapy with cetuximab, methotrexate, or docetaxel (7.5 months vs. 5.1 months; HR 0.7; p = 0.01) in patients with recurrent, platinum-resistant, squamous-cell cancer of the head and neck.
■ For the treatment of locally advanced or metastatic urothelial carcinoma, in patients with disease progression on or following platinum-containing chemotherapy, or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy.
Standard regimen
In a single-arm clinical trial (n = 270), the objective response rate of 19.6% (complete response (CR), 2.6%; partial response (PR), 17%). The median duration of response was 10.3 months (range, 1.9 months to 12+ months). Patients with PD-L1 expression of 1% or higher (n = 124) had an objective response rate of 25% (CR, 4.8%; PR, 20.2%) and those with PD-L1 expression less than 1% had an objective response rate of 15.1% (CR, 0.7%; PR, 14.4%).
■ For the treatment of microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan, as monotherapy
Adults
Standard regimen
The objective response rate (ORR) was 28% in the 53 patients who received prior fluoropyrimidine, oxaliplatin, and irinotecan. Responses lasted 6 or more months for 67% (95% CI: 38, 88) of patients. There was 1 complete response and 14 partial responses. The ORR was 32% (n = 24) (95% CI: 22, 44) among the 74 patients in the overall population.
Children and Adolescents 12 to 17 years
Standard regimen
Efficacy for pediatric patients has been extrapolated from results from clinical trials in adult MSI-H patients with additional population pharmacokinetic data.
The objective response rate (ORR) was 28% in the 53 patients who received prior fluoropyrimidine, oxaliplatin, and irinotecan. Responses lasted 6 or more months for 67% of patients. There was 1 complete response and 14 partial responses. The ORR was 32% among the 74 patients in the overall population.
■ For the treatment of microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan, in combination with ipilimumab. ........ See ipilimumab
■ For the treatment of hepatocellular cancer, after disease progression on or intolerance to sorafenib therapy.
Standard regimen
In a subgroup analysis of a multicenter, open-label clinical trial (CHECKMATE-040; n = 154), patients with hepatocellular cancer who progressed on or were intolerant to sorafenib were treated with nivolumab monotherapy. The overall response rate was 14.3%, (complete response (CR), 1.9%; partial response (PR), 12.3%. Of the 22 patients who responded to therapy, 91% had a duration of at least 6 months, while 55% maintained their response for 12 months or longer.
DOSING CONSIDERATIONS
Hepatic Impairment
Renal Impairment
CONTRAINDICATIONS / PRECAUTIONS
ADVERSE REACTIONS
DRUG INTERACTIONS
PREGNANCY AND LACTATION
FDA grants regular approval for pembrolizumab in combination with chemotherapy
for first-line treatment of metastatic nonsquamous NSCLC
On August 20, 2018, the Food and Drug Administration approved pembrolizumab (KEYTRUDA, Merck & Co., Inc.) in combination with pemetrexed and platinum as first-line treatment of patients with metastatic, non-squamous non-small cell lung cancer (NSqNSCLC), with no EGFR or ALK genomic tumor aberrations.
Pembrolizumab was previously granted accelerated approval for this indication in May 2017 based on improvements in overall response rate and progression-free survival for patients randomized to pembrolizumab administered with pemetrexed and carboplatin as compared with pemetrexed and carboplatin alone in the KEYNOTE-021 study.
Today’s approval represents fulfillment of a postmarketing commitment demonstrating the clinical benefit of this product. This action is based on the results of KEYNOTE-189 (NCT02578680), a randomized, multicenter, double-blind, active controlled study enrolling 616 patients receiving first-line treatment for metastatic NSqNSCLC. Patients were randomized (2:1) to receive pembrolizumab (or placebo) in combination with pemetrexed, and investigator’s choice of either cisplatin or carboplatin every 3 weeks for 4 cycles followed by pembrolizumab (or placebo) and pemetrexed. Treatment with pembrolizumab continued until disease progression, unacceptable toxicity, or a maximum of 24 months.
The primary efficacy outcome measures were overall survival (OS) and progression-free survival (PFS), as assessed by a blinded independent committee review (RECIST 1.1.)
The trial demonstrated a statistically significant improvement in OS for patients randomized to pembrolizumab and chemotherapy (HR 0.49; 95% CI: 0.38, 0.64; p<0.00001) in a pre-specified interim analysis. The median OS was not reached at the time of the data cut-off in the pembrolizumab plus chemotherapy arm and was 11.3 months for those in the chemotherapy arm. The trial also demonstrated an improvement in PFS for patients randomized to pembrolizumab plus chemotherapy (HR 0.52; 95% CI: 0.43, 0.64; p<0.00001). The median PFS was 8.8 months for patients receiving pembrolizumab plus chemotherapy and 4.9 months for those receiving chemotherapy alone. The overall response rate was significantly higher (48% vs. 19%; p=0.0001) for those in the pembrolizumab plus chemotherapy arm and the median response duration was 11.2 months and 7.8 months, respectively.
The most common adverse reactions reported in ≥20% of patients in KEYNOTE-189 were fatigue/asthenia, nausea, constipation, diarrhea, decreased appetite, rash, vomiting, cough, dyspnea, and pyrexia.
The recommended pembrolizumab dose and schedule for NSqNSCLC is 200 mg as an intravenous infusion over 30 minutes every 3 weeks.
Pembrolizumab (Keytruda)
Human programmed death receptor-1 (PD-1)-blocking monoclonal antibody
Approved for the treatment of certain types of melanoma, NSCLC, head and neck cancer, Hodgkin lymphoma, urothelial carcinoma, gastric (including gastroesophageal
junction) cancer, and microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors
Immune-mediated reactions have been reported; treatment may need to be withheld or permanently discontinued
Injectable Administration
Pembrolizumab is available as a single-use lyophilized powder vial (50mg) and a single-use 25 mg/mL solution vial.
Visually inspect parenteral products for particulate matter and discoloration prior to administration whenever solution and container permit.
Do not administer other drugs through the same infusion line.
Administer pembrolizumab prior to chemotherapy when given on the same day.
Lyophilized powder Reconstitution:
Add 2.3 mL of sterile water for injection, USP (SWI) to the 50 mg lyophilized powder vial for a reconstituted concentration of 25 mg/mL; inject SWI along the walls of the vial and not directly on the powder.
Gently swirl the vial and allow up to 5 minutes for bubbles to clear. Do not shake the vial. The reconstituted solution will be a clear to slightly opalescent, colorless to slightly yellow solution. Discard if visible particles are observed.
Storage following reconstitution: Store at room temperature for up to 6 hours or refrigerated at 2 to 8 degrees C (36 to 46 degrees F) for up to 24 hours (includes room temperature storage of reconstituted vials, storage of the infusion solution in the IV bag, and the duration of infusion). Do not freeze. If refrigerated, allow the diluted solution to warm to room temperature prior to administration.
Pembrolizumab single-use solution or reconstituted lyophilized powder:
Dilution:
Add the required amount of drug to a bag of 0.9% sodium chloride injection, USP or 5% dextrose injection, USP to a final diluted concentration between 1 mg/mL and 10 mg/mL. Mix by gentle inversion.
Discard any unused reconstituted solution left in the vial.
Storage following dilution: Store at room temperature for up to 6 hours or refrigerated at 2 to 8 degrees C (36 to 46 degrees F) for up to 24 hours (includes room temperature storage of reconstituted vials, storage of the diluted infusion solution, and the duration of infusion). Do not freeze. If refrigerated, allow the diluted solution to warm to room temperature prior to administration.
Intravenous Infusion:
Administer the diluted solution intravenously over 30 minutes.
Use a sterile, nonpyrogenic, low-protein binding 0.2 to 5 micron in-line or add-on filter.
STANDARD TREATMENT REGIMEN:
200 mg IV over 30 minutes repeated every 3 weeks until disease progression or up to 24 months in patients without progression. Therapy may need to be temporarily withheld or permanently discontinued in patients who develop immune-related reactions. Permanently discontinue therapy for severe or life-threatening infusion-related reactions.
■ For the treatment of unresectable or metastatic malignant melanoma.
The FDA has designated pembrolizumab as an orphan drug for the treatment of stage IIB-IV melanoma.
# In patients who have disease progression following ipilimumab or in BRAF V600 mutation-positive patients who have disease progression following ipilimumab and a BRAF inhibitor.
200 mg IV over 30 minutes repeated every 3 weeks until disease progression.
The primary endpoint of median progression-free survival (PFS) was significantly improved with pembrolizumab 2 mg/kg IV (n = 180) every 3 weeks (2.9 months) and pembrolizumab 10 mg/kg IV (n = 181) every 3 weeks (2.9 months) compared with investigator-choice chemotherapy (ICC) (n = 179; 2.7 months) in patients with unresectable stage III or IV melanoma who had disease progression following ipilimumab and had received prior treatment with a BRAF- and/or MEK-inhibitor (if BRAF V600 mutant-positive) in a second interim analysis of a multinational, randomized, phase II trial (the KEYNOTE-002 trial).
ICC consisted of carboplatin/paclitaxel (n = 42) or single-agent carboplatin (n = 13), paclitaxel (n = 28), dacarbazine (n = 45), or temozolomide (n = 43).
Ipilimumab-refractory melanoma was defined as progression within 24 weeks after at least 2 doses of ipilimumab. At a median follow-up time of 10 months, the 6-month PFS rates were 34% and 38% in the pembrolizumab 2 mg/kg and 10 mg/kg arms, respectively, compared with 16% in the ICC arm; additionally, the 9-month PFS rates were 24% and 29% compared with 8%, respectively.
Treatment crossover to pembrolizumab occurred in 48% of patients who received ICC. Overall survival (OS) data are not yet mature.
Patients with ipilimumab-refractory melanoma received pembrolizumab 2 mg/kg IV or 10 mg/kg IV every 3 weeks in a multinational, randomized, open-label, dose comparative, phase I study (n = 173; mean age, 58.8 years (range, 18 to 88 years); 3 or more prior therapies, 35%). Prior BRAF- or MEK-inhibitor therapy (or both) was required in patients with BRAF mutant melanoma (18%). At a median follow-up of 8 months, the overall response rates (ORR) were 26% in both the 2 mg/kg (n = 81) and 10 mg/kg IV (n =76) arms. The ORRs were 28% and 19% in the BRAF wild-type (n = 131) and BRAF mutant (n = 26) subgroups, respectively. The median time to response was 12 weeks in both the arms and the duration of response ranged from 6 to 37 weeks. The median PFS (22 weeks vs. 14 weeks) and OS (31 weeks vs. 35 weeks) times were not significantly different between the 2 dosing arms.
# In patients who have disease progression following no more than 1 prior therapy and have not received treatment with a CTLA-4 inhibitor or a PD-1 or PD-L1 inhibitor.
■ For the treatment of PD-L1-expressing, metastatic non-small cell lung cancer (NSCLC).
# For the first-line treatment of PD-L1-expressing (Tumor Proportion Score (TPS) 50% or higher), EGFR- and ALK-negative, metastatic non-small cell lung cancer (NSCLC), as monotherapy.
Standard regimen:
A multicenter, randomized, open-label phase 3 clinical trial was stopped early by an independent data and safety monitoring committee after demonstrating superiority of pembrolizumab compared with platinum-based chemotherapy with regard to progression-free survival (10.3 vs. 6 months) and overall survival (not reached in either group) in the first-line treatment of EGFR- and ALK- negative advanced NSCLC in patients with 50% or more PD-L1-expressing tumor cells, despite a large percentage of patients in the chemotherapy arm crossing over to receive second-line therapy with pembrolizumab.
# For the treatment of PD-L1-expressing (Tumor Proportion Score (TPS) 1% or higher), metastatic non-small cell lung cancer (NSCLC), with disease progression on or after platinum-containing chemotherapy, and after progression on EGFR- or ALK-targeted therapy if applicable, as monotherapy.
Standard regimen compared with docetaxel:
In patients with PD-L1-expressing (TPS, 1% or greater), platinum-resistant, metastatic NSCLC.
Until disease progression or up to 24 months in patients without progression.
improved overall survival (OS) and progression-free survival (PFS).
OS in patients treated with pembrolizumab was 10.4 months compared with 8.5 months in those who received docetaxel and the objective response rate (ORR) was 18% versus 9%, respectively; PFS was not significantly different.
Results were stronger in patients with TPS greater than or equal to 50%, with median OS of 14.9 months versus 8.2 months , and ORR 30% compared with 8%, respectively; the improvement in PFS was small but statistically significant in this subgroup (5.2 months vs. 4.1 months).
# For the first-line treatment of metastatic nonsquamous non-small cell lung cancer (NSCLC), in combination with pemetrexed and carboplatin. (with no EGFR or ALK genomic tumor aberrations)
200 mg IV over 30 minutes on day 1, followed by pemetrexed 500 mg/m2 IV over 10 minutes plus carboplatin AUC 5 IV infusion on day 1, every 3 weeks for 4 cycles.
Premedicate pemetrexed with dexamethasone 4 mg by mouth twice daily for 3 days, beginning the day before pemetrexed administration to reduce cutaneous reactions.
Additionally, supplement with folic acid (400 to 1,000 mcg by mouth daily) and vitamin B12 (1 mg IM every 3 cycles) beginning 7 days prior to the first dose of pemetrexed and continuing for 21 days after the last dose to reduce the severity and frequency of hematologic and GI toxicities; after the first dose, vitamin B12 may be given on the same day as pemetrexed. Do not substitute oral for IM vitamin B12.
After completion of 4 cycles of pembrolizumab, pemetrexed, and carboplatin, continue pembrolizumab standard regimen for up to a maximum of 24 months.
In a multicenter, open-label clinical trial (KEYNOTE-021), patients with previously untreated, locally advanced or metastatic nonsquamous NSCLC who received treatment with pembrolizumab, pemetrexed, and carboplatin (n = 60) had a statistically significant improvement in overall response rate (ORR) compared with pemetrexed and carboplatin alone (n = 63) (55% vs. 29%), with the response duration lasting over 6 months in 93% of patients in the pembrolizumab group compared with 81% in the chemotherapy alone group. The median progression-free survival was 13 months compared with 8.9 months, respectively. In an exploratory analysis, patients with less than 1% PD-L1 expression in tumor cells, the ORR was 57% in the pembrolizumab arm and 13% in the chemotherapy arm; in patients with greater than or equal to 1% PD-L1 expression, the ORR was 54% and 38%, respectively.
■ For the treatment of recurrent or metastatic head and neck cancer (squamous cell) with disease progression on or after platinum-containing chemotherapy.
Standard regimen:
After a median follow-up of 8.9 months, treatment with pembrolizumab resulted in an objective response rate (ORR) of 16% with complete response rate of 5%.
The median duration of response was not reached, but ranged from 2.4 months to more than 27.7 months; 23 of 28 responses lasted 6 months or longer. The ORR and duration of response were similar regardless of dosage regimen (10 mg/kg every 2 weeks or 200 mg every 3 weeks) or HPV status.
■ For the treatment of Hodgkin's disease
† For the treatment of classical Hodgkin's disease that has relapsed or progressed after an autologous stem-cell transplant and post-transplant brentuximab vedotin.
Standard regimen:
In a phase II trial (KEYNOTE-087 trial).
# For the treatment of classical Hodgkin's disease in patients with refractory disease or who have relapsed after 3 or more prior lines of therapy.
Standard regimen:
The overall response rate was 69% in patients with relapsed or refractory classical Hodgkin lymphoma who received pembrolizumab in a multicenter, nonrandomized trial (n = 210; Keynote-087). Additionally, a complete remission was achieved in 22% of patients.
At a median follow-up time of 9.4 months, the median duration of response was 11.1 months. Patients (median age, 18 to 76 years) in this study had received a median of 4 prior therapies (range, 1 to 12 therapies); 61% of patients had previously received an autologous stem-cell transplant, 83% of patients had received prior brentuximab therapy, and 36% of patients had received prior radiation therapy.
Adolescents, Children, and Infants
2 mg/kg (not to exceed 200 mg) IV over 30 minutes repeated every 3 weeks until disease progression or up to 24 months.
Efficacy for pediatric patients has been extrapolated from results from clinical trials in adult classical Hodgkin lymphoma patients.
■ For the treatment of locally advanced or metastatic urothelial carcinoma.
# For the treatment of locally advanced or metastatic urothelial carcinoma in patients who progress during or following platinum-containing chemotherapy.
Standard regimen compared with investigator’s choice of chemotherapy:
Treatment with pembrolizumab significantly improved median overall survival (10.3 vs. 7.4 months) and objective response rates (21% vs. 11%; complete response, 7% vs. 3%; partial response, 14% vs. 8%).
Investigator’s choice of chemotherapy (paclitaxel, docetaxel, vinflunine).
The median duration of response was not reached in the pembrolizumab arm, compared with 4.3 months in the chemotherapy arm. There was no significant difference in progression-free survival between treatment arms.
# For the treatment of locally advanced or metastatic urothelial carcinoma in patients who progress within 12 months of neoadjuvant or adjuvant platinum-based chemotherapy.
Standard regimen compared with investigator’s choice of chemotherapy:
In a multicenter, randomized clinical trial (KEYNOTE-045), treatment with pembrolizumab significantly improved median overall survival (10.3 vs. 7.4 months) and objective response rates (21% vs. 11%; complete response, 7% vs. 3%; partial response, 14% vs. 8%).
Investigator’s choice of chemotherapy (paclitaxel, docetaxel, vinflunine)
The median duration of response was not reached in the pembrolizumab arm, compared with 4.3 months in the chemotherapy arm. There was no significant difference in progression-free survival between treatment arms.
Fifteen percent of patients in this trial had disease progression following platinum-based neoadjuvant or adjuvant chemotherapy.
# For the treatment of locally advanced or metastatic urothelial carcinoma in patients who are not eligible for cisplatin-containing chemotherapy.
Standard regimen:
In a multicenter, open-label, single-arm clinical trial, treatment with pembrolizumab resulted in an objective response rate of 29% with 7% complete responses and 22% partial responses. The median duration of response was not reached.
■ For the treatment of unresectable or metastatic microsatellite instability-high solid tumors or mismatch repair deficient solid tumors.
# For the treatment of unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) colorectal cancer that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
Standard regimen:
In pooled data from 5 multicenter, uncontrolled, open-label, multicohort, single-arm trials (KEYNOTE-016, KEYNOTE-164, KEYNOTE-028, KEYNOTE-012, and KEYNOTE-158), patients with MSI-H or dMMR colorectal cancer treated with pembrolizumab (10 mg/kg every 2 weeks or 200 mg every 3 weeks; n = 90) had an objective response rate of 36% (95% CI, 26% to 46%) with a median duration of response ranging from 1.6+ to 22.7+ months.
Children and Adolescents 2 to 17 years
2 mg/kg (not to exceed 200 mg) IV over 30 minutes repeated every 3 weeks until disease progression or up to 24 months in patients without progression.
Efficacy for pediatric patients has been extrapolated from results from clinical trials in adult MSI-H patients. In pooled data from 5 multicenter, uncontrolled, open-label, multicohort, single-arm trials, adult patients with MSI-H or dMMR colorectal treated with pembrolizumab (10 mg/kg every 2 weeks or 200 mg every 3 weeks; n = 90) had an objective response rate of 36% (95% CI, 26% to 46%) with a median duration of response ranging from 1.6+ to 22.7+ months.
# For the treatment of unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and have no satisfactory alternative treatment options.
Standard regimen:
In pooled data from 5 multicenter, uncontrolled, open-label, multicohort, single-arm trials, patients with MSI-H or dMMR solid tumors treated with pembrolizumab (10 mg/kg every 2 weeks or 200 mg every 3 weeks; n = 149) had an objective response rate of 39.6% (complete response, 7.4%; partial response, 32.2%) with a median duration of response not reached (range, 1.6+ to 22.7+ months); 78% of patients had a duration of response greater than or equal to 6 months.
Children and Adolescents 2 to 17 years
2 mg/kg (not to exceed 200 mg) IV over 30 minutes repeated every 3 weeks until disease progression or up to 24 months.
Efficacy for pediatric patients has been extrapolated from results from clinical trials in adult MSI-H patients. In pooled data from 5 multicenter, uncontrolled, open-label, multicohort, single-arm trials, adult patients with MSI-H or dMMR solid tumors treated with pembrolizumab (10 mg/kg every 2 weeks or 200 mg every 3 weeks; n = 149) had an objective response rate of 39.6% (complete response, 7.4%; partial response, 32.2%) with a median duration of response not reached (range, 1.6+ to 22.7+ months); 78% of patients had a duration of response greater than or equal to 6 months.
■ For the treatment of recurrent, PD-L1 positive (combined positive score [CPS] 1 or higher), locally advanced or metastatic gastric cancer, including gastroesophageal junction adenocarcinoma (GEJ), with disease progression on or after 2 or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
Standard regimen:
In a multicenter, open-label mult-cohort trial of patients with gastric or GEJ adenocarcinoma who progressed on at least 2 prior systemic treatments for advanced disease, the objective response rate of patients treated with pembrolizumab was 13.3% (95% CI, 8.2% to 20%); 1.4% had a complete response (CR) and 11.9% had a partial response (PR). For responding patients, the duration of response ranged from 2.8 to 19.4+ months; 58% of responding patients had a duration of response of at least 6 months and 26% had responses of 12 months or longer. Four (4) of 7 patients with microsatellite instability-high (MSI-H) tumors had objective responses, including one CR; the duration of response in these patients ranged from 5.3 months to 14.1+ months.
† For the treatment of Merkel cell carcinoma.
† For the treatment of recurrent locoregional or metastatic Merkel cell carcinoma in patients who had progressive disease following standard therapy with surgery and/or radiation therapy.
2 mg/kg IV repeated every 3 weeks until disease progression or up to a maximum of 24 months.
(Median duration of treatment 27 weeks; range, 3 to 57 weeks) was evaluated in a phase II study (n = 25).
Patients who achieved a complete response (CR) could discontinue pembrolizumab if they had at least 6 months of therapy and received at least 2 cycles of therapy after a confirmed CR.
† Indicates off-label use
bcr-abl tyrosine kinase inhibitors
BCR-ABL signalling pathways.

Bcr-Abl Inhibitors
| Inhibitor Name | Bcr-Abl | Abl | Other Targets |
| Imatinib Mesylate | + | c-Kit,PDGFR | |
| Dasatinib Monohydrate | ++++ | ++++ | Src,c-Kit (D816V),c-Kit (wt) |
| Nilotinib | ++ |
Imatinib Mesylate
US Brand Name: Gleevec
FDA Approved: Yes
The mesylate salt of imatinib, a tyrosine kinase inhibitor with antineoplastic activity. Imatinib binds to an intracellular pocket located within tyrosine kinases (TK), thereby inhibiting ATP binding and preventing phosphorylation and the subsequent activation of growth receptors and their downstream signal transduction pathways. This agent inhibits TK encoded by the bcr-abl oncogene as well as receptor TKs encoded by the c-kit and platelet-derived growth factor receptor (PDGFR) oncogenes. Inhibition of the bcr-abl TK results in decreased proliferation and enhanced apoptosis in malignant cells of Philadelphia-positive (Ph+) hematological malignancies such as CML and ALL; effects on c-kit TK activity inhibit mast-cell and cellular proliferation in those diseases overexpressing c-kit, such as mastocytosis and gastrointestinal stromal tumor (GIST). Check for active clinical trials using this agent. (NCI Thesaurus)
Chemical structure: 4-[(4-methyl-1-piperazinyl)methyl]-N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]-phenyl]benzamide methanesulfonate
Use in Cancer - Imatinib mesylate is approved to treat:
• Acute lymphoblastic leukemia in adults and children that is Philadelphia chromosome positive. In adults, it is used for disease that has recurred (come back) or is refractory (does not respond to treatment). In children, it is used as the first treatment after the disease is diagnosed.
• Chronic eosinophilic leukemia or hypereosinophilic syndrome.
• Chronic myelogenous leukemia that is Philadelphia chromosome positive.
• Dermatofibrosarcoma protuberans.
• Gastrointestinal stromal tumor (GIST).
• Myelodysplastic/myeloproliferative neoplasms.
• Systemic mastocytosis.
Imatinib Mesylate
GLEEVEC tablets are indicated for:
• Newly diagnosed adult and pediatric patients with Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in the chronic phase
• Patients with Ph+ CML in blast crisis (BC), accelerated phase (AP), or in the chronic phase (CP) after failure of interferon-alpha therapy
• Adult patients with relapsed or refractory Ph+ acute lymphoblastic leukemia (Ph+ ALL)
• Pediatric patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) in combination with chemotherapy
• Adult patients with myelodysplastic/myeloproliferative diseases (MDS/MPD) associated with PDGFR (platelet-derived growth factor receptor) gene rearrangements as determined with an FDA-approved test
• Adult patients with aggressive systemic mastocytosis (ASM) without the D816V c-KIT mutation as determined with an FDA-approved test or with c-KIT mutational status unknown
• Adult patients with hypereosinophilic syndrome (HES) and/or chronic eosinophilic leukemia (CEL) who have the FIP1L1-PDGFRα fusion kinase and for patients with HES and/or CEL who are FIP1L1-PDGFRα fusion kinase negative or unknown
• Adult patients with unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans (DFSP)
• Patients with KIT (CD117)-positive gastrointestinal stromal tumors (GIST) that cannot be surgically removed and/or have spread to other parts of the body
• Adult patients after surgery who have had their KIT (CD117)-positive GIST completely removed
Imatinib Mesylate
An oral tyrosine kinase inhibitor that blocks the receptors for BCR-ABL, platelet-derived growth factor (PDGF), stem cell factor (SCF), and c-kit
Used for Philadelphia chromosome-positive chronic myeloid leukemia and acute lymphoblastic leukemia, myelodysplastic/myeloproliferative diseases, mastocytosis, hypereosinophilic syndrome and/or chronic eosinophilic leukemia, and c-kit (CD117)-positive gastrointestinal stromal tumors
Serious hepatotoxicity has occurred; monitoring is required
HOW SUPPLIED
Gleevec/Imatinib/Imatinib Mesylate Oral Tab: 100mg, 400mg
DOSAGE & INDICATIONS:
■ For the treatment of Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia (CML).
# For the treatment of newly diagnosed chronic-phase Ph+ CML.
400 mg PO once daily with food. Treatment is continued until disease progression or unacceptable toxicity.
Avoid concomitant use of strong CYP3A4 inducers if possible; increase the imatinib dosage if concomitant use is necessary.
Consider increasing to 600 mg PO once daily in the absence of severe adverse reactions and severe non-leukemia related neutropenia or thrombocytopenia in the following circumstances:
disease progression (at any time);
failure to achieve a satisfactory hematologic response after at least 3 months of treatment;
loss of a previously achieved hematologic response or cytogenetic response; or
failure to achieve a cytogenetic response after 6 to 12 months of treatment.
In patients with newly diagnosed chronic-phase chronic myelogenous leukemia (CML), the complete cytogenic response (CCyR) rate was 74.7% in patients who received oral imatinib (n = 553) compared with 6.5% in patients who received subcutaneous cytarabine plus interferon (n = 553) in a multinational, randomized, phase III study.
Imatinib dosage escalation (up to 800 mg/day) was permitted. The estimated progression-free survival rate at 30 months was 87.8% in the imatinib arm and 68.3% in the combination arm (p < 0.001).
After a median follow-up of 60 months, 65% of patients in the cytarabine plus interferon crossed over to the imatinib arm;
subsequent analyses of the patients in this trial focused only on those originally randomized to imatinib. The CCyR rate was 87%, the overall survival (OS) rate was 89%, and 93% of patients had not progressed to blast crisis or to the accelerated phase.
The primary endpoint of major molecular response (MMR) rate at 12 months was significantly improved with nilotinib 300 mg twice daily (44%) and nilotinib 400 mg twice daily (43%) compared with imatinib 400 mg once daily (22%; p < 0.001 for both comparisons) in patients with newly diagnosed chronic phase Philadelphia chromosome-positive CML in a multicenter, randomized, phase III trial (the ENESTnd trial; n = 846).
Additionally, the 12-month CCyR rates were significantly higher for nilotinib (600 mg/day, 80%; 800 mg/day, 78%) compared with imatinib (65%; p < 0.001 for both comparisons).
At a minimum follow-up of 24 months, the 24-month MMR rates continued to be significantly higher for nilotinib (600 mg/day, 71%; 800 mg/day, 67%) compared with imatinib (44%; p < 0.0001 for both comparisons). At a follow-up of 60 months, the median OS time had not been reached in any study arm; the estimated OS rate was 93.7% in the nilotinib 600-mg/day arm and 91.7% in the imatinib arm. Additionally, the MMR rate continued to be higher in patients who received nilotinib therapy (77% vs. 60%).
Children >= 1 year and Adolescents
340 mg/m2/day orally; do not exceed 600 mg/day. The daily dose may be given as a single dose or split into 2 doses given once in the morning and once in the evening. Treatment is continued until disease progression or unacceptable toxicity. Avoid concomitant use of strong CYP3A4 inducers if possible. Treatment with imatinib led to an 8-week complete hematologic response rate of 78% and a complete cytogenetic response rate of 65% in pediatric patients with chronic phase, Philadelphia chromosome-positive chronic myelogenous leukemia in a multicenter, single-arm, phase II trial (n = 51).
# For the treatment of chronic-phase Ph+ CML after the failure of interferon-alfa therapy.
400 mg PO once daily. Consider increasing to 600 mg PO once daily in the absence of severe adverse reactions and severe non-leukemia related neutropenia or thrombocytopenia in the following circumstances:
disease progression (at any time);
failure to achieve a satisfactory hematologic response after at least 3 months;
loss of a previously achieved hematologic response.
Treatment is continued until disease progression or unacceptable toxicity.
Avoid concomitant use of strong CYP3A4 inducers if possible; increase the imatinib dosage if concomitant use is necessary.
Patients with chronic phase CML who have failed interferon alfa therapy demonstrated a hematologic response rate of 95%, with 60% of patients achieving a major cytogenetic response. Thirty-nine percent achieved a complete cytogenetic response confirmed by a second bone marrow cytogenetic evaluation performed at least one month after the initial bone marrow study.
The median time to hematologic response was 1 month. In one report, patients with chronic phase CML post-interferon failure were treated with imatinib 400 mg PO twice daily. Of evaluable patients, 19/21 (90%) achieved a major cytogenetic response. Toxicities were similar to those reported with standard dose.
# For pediatric patients with chronic phase Ph+ CML whose disease has recurred after hematopoietic stem cell transplant or who are resistant to interferon-alfa therapy†.
Children >= 3 years, Adolescents, and Adults <= 20 years
260 mg/m2/day PO given as a single daily dose or the dose may be divided given once in the morning and once in the evening.
Consider increasing to 340 mg/m2/day PO in the absence of severe adverse reactions and severe non-leukemia related neutropenia or thrombocytopenia in the following circumstances:
disease progression (at any time);
failure to achieve a satisfactory hematologic response after at least 3 months of treatment;
loss of a previously achieved hematologic response or cytogenetic response; or
failure to achieve a cytogenetic response after 6 to 12 months of treatment.
In an open-label single-arm study, 14 pediatric patients with Ph+ chronic phase CML recurrent after stem cell transplant or resistant to interferon alfa therapy were treated with imatinib. In the 13 patients for whom cytogenetic information is available, 4 achieved a major cytogenetic response, 7 achieved a complete cytogenetic response, and 2 had minimal cytogenetic response. The cytogenetic response rate was similar at all dosage levels studied.
# For the treatment of accelerated-phase or blast crisis Ph+ CML after the failure of interferon-alfa therapy.
600 mg PO once daily.
Consider increasing to 800 mg/day PO (400 mg PO twice daily) in the absence of severe adverse reactions and severe non-leukemia related neutropenia or thrombocytopenia in the following circumstances:
disease progression (at any time);
failure to achieve a satisfactory hematologic response after at least 3 months of treatment;
loss of a previously achieved hematologic response or cytogenetic response; or
failure to achieve a cytogenetic response after 6 to 12 months of treatment. Treatment is continued until disease progression or unacceptable toxicity.
Avoid concomitant use of strong CYP3A4 inducers if possible; increase the imatinib dosage if concomitant use is necessary.
In patients with CML in accelerated phase or blast crisis, the hematologic response (doses 400 to 600 mg/day) was 69% and 52%, respectively, with major cytogenic responses in 21% and 13.5%, respectively. Twenty-four percent of patients in accelerated phase and 19% in blast crisis returned to chronic phase CML. In blast crisis, the median duration of hematologic response is about 7 months, and in accelerated phase, the median duration is more than 6 months, but the specific median duration of response cannot yet be estimated.
■ For the treatment of Philadelphia chromosome-positive (Ph+) acute lymphocytic leukemia (ALL).
# For the treatment of relapsed or refractory Ph+ ALL.
600 mg orally once daily. Treatment is continued until disease progression or unacceptable toxicity.
Avoid concomitant use of strong CYP3A4 inducers if possible; increase the imatinib dosage if concomitant use is necessary.
Single-agent imatinib therapy (median duration of 62 days; range, 14 to 343 days) resulted in a sustained hematologic response (lasting at least 4 weeks) in 27% of relapsed or refractory, Philadelphia chromosome-positive acute lymphocytic leukemia patients in a multicenter, phase II trial (n = 48; prior bone morrow transplant, n = 10 (21%)); a sustained complete hematologic response (CHR) was achieved in 6% of patients. Additionally, 17% of patients had a complete cytogenetic response. The estimated median time to progression was 2.2 months and the estimated median overall survival time was 4.9 months.
# For the treatment of newly diagnosed Ph+ ALL, in combination with chemotherapy.
Children >= 1 year, Adolescents, and Adults <= 21 years
340 mg/m2 orally once daily; do not exceed 600 mg/day. Avoid concomitant use of strong CYP3A4 inducers if possible.
The 3-year event-free survival (EFS) rate was 80.5% +/- 11.2% (95% CI, 64.5%—89.8%) in a cohort of 50 patients aged 1 to 21 years (median age, 10 years) with very high-risk Philadelphia chromosome-positive acute lymphocytic leukemia (ALL) who received imatinib after 4 to 6 weeks of 3- or 4-drug induction therapy in a multicenter, nonrandomized trial (the COG AALL0031 trial). Most patients in this cohort (n = 44) received continuous imatinib therapy for 280 days prior to maintenance. High-risk ALL was defined as patients with an expected 5-year EFS of less than 45% with conventional chemotherapy. In this trial, patients received daily imatinib starting with the first course of post-induction chemotherapy and continued through maintenance chemotherapy cycles 1 to 4; imatinib was administered intermittently on a 2-week-on/2-week-off schedule during maintenance cycles 5 to 12. The 20 patients who underwent hematopoietic stem cell transplant (HSCT) received 42 days of imatinib prior to HSCT and 28 weeks (196 days) of imatinib after the immediate post transplant period. At a median follow-up time of 40.5 months, the estimated 4-year EFS of patients in this study was 70% (95% CI, 54% to 81%).
■ For the treatment of Kit (CD117) positive gastrointestinal stromal tumors (GIST).
# For the treatment of Kit (CD117) positive unresectable and/or metastatic GIST.
400 mg by mouth once daily until disease progression or unacceptable toxicity.
If patients show clear signs or symptoms of disease progression at a lower dose and in the absence of severe adverse drug reactions, the dose may be increased up to 800 mg per day (given as 400 mg twice daily) as clinically indicated; however, the incidence of anemia and asthenia increases significantly at this dosage and the median time to progression after dose escalation is approximately 80 days.
Coadministration of certain drugs may need to be avoided or dosage adjustments may be necessary; review drug interactions.
In 2 multicenter, randomized, open-label clinical trials (n = 1,640), patients with unresectable or metastatic GIST were randomized to treatment with either 400 mg or 800 mg per day of imatinib; patients in the 400 mg arm were permitted to cross over to receive 800 mg daily upon disease progression. The primary outcome of progression-free survival (PFS) was 18.9 months compared with 23.2 months, respectively. Partial responses occurred in 46.1% of the 400-mg arm compared with 48.9% of the 800-mg arm, while complete responses were recorded in 5.3% versus 5% of patients, respectively. There were no differences in overall survival between treatment groups (p = 0.98).
In another multicenter, open-label phase 2 clinical trial, there were no differences in response rates between patients treated with either 400 mg or 600 mg of imatinib per day.
# For the adjuvant treatment of Kit (CD117) positive GIST after complete gross resection.
400 mg by mouth once daily. Coadministration of certain drugs may need to be avoided or dosage adjustments may be necessary; review drug interactions.
A treatment duration of 3 years in patients with tumor diameter greater than 5 cm and mitotic count greater than 5/50 high power fields (HPF), tumor diameter greater than 10 cam and any mitotic count, any tumor size with mitotic count greater than 10/50 HPF, or tumors ruptured into the peritoneal cavity.
The optimal duration of treatment for other patients is unknown, but in clinical trials durations of both 1 year and 3 years have been studied. In a planned 15-month interim analysis of a multicenter, randomized, double-blind clinical trial, recurrence-free survival (RFS) was improved with 1-year of imatinib treatment (n = 359) compared with placebo (n = 354) (HR 0.398; p less than 0.0001). Twenty percent of placebo arm crossed over to receive imatinib after the interim analysis, and the updated RFS hazard ratio after 50 months was 0.718 (95% CI, 0.531 to 0.971). Overall survival was not significantly improved by adjuvant imatinib. In a second multicenter, randomized, open-label phase 3 clinical trial, 36 months of imatinib treatment significantly prolonged RFS compared with 12 months of imatinib treatment (HR 0.46; p less than 0.0001) in patients with tumor diameter greater than 5 cm with mitotic count greater than 5/50 high power fields (HPF), tumor diameter greater than 10 cam with any mitotic count, any tumor size with mitotic count greater than 10/50 HPF, or tumors ruptured into the peritoneal cavity. Overall survival was also significantly improved in the 36-month treatment arm (HR 0.45; p = 0.0187).
■ For the treatment of hypereosinophilic syndrome (HES) and/or chronic eosinophilic leukemia (CEL) who have the FIPL1L1-PDGFR alpha fusion kinase (mutational analysis or FISH demonstration of CHIC2 allele deletion) and for patients with HES and/or CEL who are FIPL1L-PDGFR alpha fusion kinase negative or unknown.
400 mg PO once daily in patients who are FIPL1L-PDGFR alpha fusion kinase negative or unknown.
For HES/CEL patients with demonstrated FIP1L1-PDGFR alpha fusion kinase, a starting dose of 100 mg/day PO is recommended. Increase dose from 100 mg to 400 mg for these patients may be considered in those who have had an inadequate response to therapy in the absence of adverse drug reactions.
Treatment should continue as long as the patient continues to benefit. Several small series of patients with HES have reported responses to imatinib at dosages of 100 to 400 mg/day PO.
In one series, 9 of 11 patients treated with imatinib had responses lasting more than 3 months in which the eosinophil count returned to normal. In another series, patients whose disease did not respond to imatinib 100 mg/day PO after 4 weeks had their dosage increased to 400 mg/day PO. In 3 of the 4 without response, the disease failed to respond to an increase in dosage. The dosage of imatinib should be increased by at least 50% and clinical response closely monitored in patients receiving imatinib with a potent cytochrome P450 inducer such as rifampin or phenytoin.
■ For the treatment of myelodysplastic syndrome (MDS)/myeloproliferative disease (MPD) associated with the PDGFR (platelet-derived growth factor receptor) gene rearrangements.
Prior to starting therapy, test for platelet-derived growth factor receptor (PDGFR) gene re-arrangements using a FDA-approved test.
400 mg PO once daily. Treatment should continue as long as the patient continues to benefit.
An open label phase 2 trial of patients with life-threatening conditions associated with Abl, Kit, or PDGFR protein tyrosine kinases included 7 patients with MDS/MPD who were treated with imatinib 400 mg/day. In addition, case reports and series describe an additional 24 patients with MDS/MPD treated with imatinib 400 mg/day. Of the total patients treated (n = 31), 14 (45%) achieved a complete hematologic response and 12 (39%) a major cytogenetic response (including 10 with a complete cytogenetic response). Response durations in the phase 2 trial ranged from 141+ days to 457+ days; median duration of response was 12.9 months.
■ For the treatment of aggressive systemic mastocytosis (ASM) without D816V c-Kit mutation or with c-Kit mutation status unknown.
Prior to starting therapy, test for D816V c-Kit mutation status using a FDA-approved test.
400 mg PO once daily in patients without the FIP1L1-PDGFR alpha c-Kit mutation.
If c-Kit status is unknown or not available, treatment with 400 mg PO once daily may be considered for patients with ASM not responding to other therapies.
For patients with ASM associated with eosinophilia, a starting dose of 100 mg/day PO is recommended. Dose increases from 100 mg to 400 mg for these patients may be considered in those who have had an inadequate response to therapy the absence of adverse drug reactions. Treatment should continue as long as the patient continues to benefit.
An open label phase 2 trial of patients with life-threatening conditions associated with Abl, Kit, or PDGFR protein tyrosine kinases included 5 patients with ASM who were treated with imatinib 100 to 400 mg/day. In addition, case reports and series describe 23 other patients with ASM treated with imatinib 100 to 400 mg/day. Of the total patients treated (n = 28), 8 (29%) achieved a complete hematologic response and 9 (32%) a partial hematologic response (61% overall response rate). In the literature, the response duration ranged from 1+ to 30+ months.
■ For the treatment of unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans (DFSP).
NOTE: The dosage of imatinib should be increased by at least 50% and clinical response closely monitored in patients receiving imatinib with a potent cytochrome P450 inducer such as rifampin or phenytoin (see Drug Interactions).
400 mg PO twice daily (800 mg/day). Treatment should continue as long as the patient continues to benefit.
An open label phase 2 trial of patients with life-threatening conditions associated with Abl, Kit, or PDGFR protein tyrosine kinases included 12 patients with DFSP who were treated with imatinib 800 mg/day. In addition, case reports and series describe 6 other patients with DFSP treated with imatinib either 400 or 800 mg/day. A single pediatric patient received 400 mg/m2/day increased to 520 mg/m2/day. Of the total 18 patients, 8 of them had metastatic disease. Response in those with DFSP included 7 (39%) complete responses and 8 (44%) partial responses with an overall response rate of 83%.
■ For the treatment of desmoid tumor† or aggressive fibromatosis† not amenable to surgery or radiotherapy.
Various dosage regimens ranging from 200 to 800 mg/day PO have been studied. 400 mg/day PO for up to 12 months (increased to 800 mg/day if disease progression occurred) resulted in a 3-month response rate (complete response [CR] + partial response [PR] + stable disease [SD]) of 91% (CR, 2.9%; PR, 8.6%) and a median progression-free survival time of 25 months in 35 evaluable patients in a phase II study. One patient in this study experienced grade 3 rhabdomyolysis. In another phase II study, the 4-month clinical benefit rate was 84% in 51 patients aged 12 to 67 years with AF who received imatinib 100 mg PO twice daily (BSA less than 1 m2), 200 mg PO twice daily (BSA 1 to 1.4 m2), or 300 mg PO twice daily (BSA more than 1.5 m2); additionally, 3 patients had a PR after 19, 22, and 26 months of therapy. In a phase II study in 19 heavily-pretreated patients, 800 mg/day PO resulted in 3 partial responses (15.7%) lasting greater than 1.5 years (range, 594 to 1494+ days), 4 patients with stable disease lasting greater than 1 year, and a median time to progression of 325 days. Dose reductions to 400 to 600 mg/day PO were required for most patients due to grade 3 or higher toxicity.
Children >= 12 years and Adolescents
100 mg PO twice daily (BSA less than 1 m2), 200 mg PO twice daily (BSA 1 to 1.4 m2), or 300 mg PO twice daily (BSA more than 1.5 m2) has been studied in a phase II study. The 4-month clinical benefit rate was 84% in 51 patients; additionally, 3 patients had a PR after 19, 22, and 26 months of therapy.
Targeted therapy with tyrosine kinase inhibitors
A trial randomly assigning 1,106 previously untreated patients to imatinib mesylate or to interferon plus cytarabine documented a 76% complete cytogenetic response rate with imatinib mesylate versus 14% for interferon plus cytarabine at a median follow-up of 19 months.[Level of evidence: 1iiDiii]
At 18 months, 96.7% of the imatinib group had avoided progression to accelerated-phase chronic myelogenous leukemia (CML) or blast crisis compared with 91.5% of the interferon plus cytarabine group (P < .001).
Because 90% of the combination group had switched to imatinib by 18 months (mostly because of intolerance of side effects), a survival difference may never be observed.
By the 5-year median follow-up of this trial, imatinib mesylate induced complete cytogenetic response in more than 80% of the participants, with the annual rate of progression to accelerated-phase CML or blast crisis dropping from 2% in the first year to less than 1% in the fourth year. In addition, the overall survival (OS) rate for all patients at 5 years is 89%, with fewer than 50% of all deaths (4.5%) caused by CML. More than 90% of completely responding patients still show detectable evidence of the BCR/ABL translocation, usually by reverse transcription-polymerase chain reaction (RT–PCR) or by fluorescence in situ hybridization of progenitor cell cultures. Poor compliance is the predominant reason for inadequate molecular response to imatinib.
Tyrosine kinase inhibitors with greater potency and selectivity for BCR/ABL than imatinib have been evaluated in newly diagnosed patients with CML.
In a randomized, prospective study of 846 patients that compared nilotinib with imatinib, the rate of major molecular response at 24 months was 71% and 67% for two-dose schedules of nilotinib and 44% for imatinib (P < .0001 for both comparisons).[Level of evidence: 1iiDiv] Progression to accelerated-phase CML or blast crisis occurred in 17 patients on imatinib (14%), but this progression only occurred in two patients (<1%, P = .0003) and in five patients (1.8%, P = .0089), respectively, for those patients on two-dose schedules of nilotinib. Nilotinib-treated patients had a lower rate of treatment-emergent BCR/ABL mutations than did imatinib-treated patients.
Similarly, in a randomized, prospective study of 519 patients that compared dasatinib with imatinib, the rate of major molecular response at 12 months was 46% for dasatinib and 28% for imatinib (P < .0001). The rate of major molecular response at 24 months was 64% for dasatinib and 46% for imatinib (P < .0001).[Level of evidence: 1iiDiv] At 5 years, there was no difference in progression-free survival (PFS) or OS. Progression to accelerated-phase CML or blast crisis occurred in 13 patients (5%) on imatinib and in 6 patients (2.3%) on dasatinib (not statistically different).
Although one of these two studies showed statistically significant decreased rates of progression to accelerated- or blastic-phase CML, the 5- to 10-year follow-up period with nilotinib and dasatinib demonstrated a similar survival for these agents, similar to that for imatinib. In randomized prospective trials, nilotinib and dasatinib show higher rates of earlier molecular response compared with imatinib; whether this will translate to improved long-term outcomes remains unclear.[Level of evidence: 1iiDiv] The preferred initial treatment for newly diagnosed patients with chronic-phase CML could be any of these specific inhibitors of the BCR/ABL tyrosine kinase.
A BCR/ABL transcript level of less than 10% in patients after 3 months of treatment with a specific tyrosine kinase inhibitor is associated with the best prognosis in terms of failure-free survival, PFS, and OS. However, in a retrospective analysis, even patients with a BCR/ABL transcript level greater than 10% after 3 months of therapy did well when the halving time was less than 76 days. Mandating a change of therapy based on this 10% transcript level at 3 to 6 months is problematic because 75% of patients do well even with a suboptimal response.
Higher doses of imatinib mesylate, alternative tyrosine kinase inhibitors (such as dasatinib or nilotinib, and allogeneic SCT) are implemented for suboptimal response or progression and are under clinical evaluation as front-line approaches. Dose escalation of imatinib can be considered for patients with suboptimal response, but clinical trials are required to establish the relative efficacy and sequencing of dose escalation versus the use of dasatinib or nilotinib. Two studies looked at dose escalation of imatinib in almost 200 previously untreated patients, most of whom were of intermediate Sokal risk; 63% to 73% achieved a major molecular response by 18 to 24 months and only three patients showed progression to advanced phase in these preliminary phase II results.[Level of evidence: 3iiiDiv] Until randomized studies are performed, it is unclear whether the increased response with increased dosage will translate into longer durations of response or survival advantages.
A single-arm clinical trial using first-line imatinib with either selective imatinib intensification or selective switching to nilotinib resulted in a 3-year OS of 96% and transformation-free survival of 95%, with a confirmed major molecular response rate of 73% at 24 months.[Level of evidence: 3iiiDiv] All patients started treatment with imatinib and were given 600 mg daily. Imatinib plasma trough levels that were under 1,000 ng/mL on day 22 prompted an increase of imatinib to 800 mg daily (20% of patients). Molecular targets were set, and failure to reach these targets prompted an increase of imatinib to 800 mg daily (if not already performed) or a switch to nilotinib. The molecular targets were as follows:
3 months: BCR-ABL ≤ 10%.
6 months: BCR-ABL ≤ 1%.
12 months: BCR-ABL ≤ 0.1%.
This strategy of employing front-line imatinib is an alternative to the immediate use of more-potent tyrosine kinase inhibitors, such as nilotinib and dasatinib.
A single-center, retrospective analysis of 483 patients with chronic-phase CML who were treated with imatinib (400 mg or 800 mg qd), dasatinib, or nilotinib indicated that patients who have better than 35% t(9;22)+ cells at 3 months of therapy have inferior event-free, transformation-free, and OS rates compared with patients who have better early cytogenetic responses.
Among the many unanswered questions are the following:
• Should the newer tyrosine kinase inhibitors dasatinib and nilotinib replace imatinib as front-line therapy? Randomized trials have failed to confirm OS differences. Imatinib blood levels and timed molecular targets that informed the need for increased doses of imatinib may make any clinical differences between nilotinib, dasatinib, and imatinib more about side effects than about efficacy.
• Does time-to-response matter if a good response is obtained eventually?
• Does a good response in a high-risk patient overcome the adverse prognosis of the high-risk features?
• Should other active agents be added to therapy with tyrosine kinase inhibitors?
All of these issues have led to an active reappraisal of recommendations for optimal front-line therapy for chronic-phase CML.
For patients who obtain a complete molecular remission, the question is whether therapy with tyrosine kinase inhibitors can be discontinued. A review of several retrospective reports can be summarized as follows:[Level of evidence: 3iiiDiv]
1. Patients who have taken a tyrosine kinase inhibitor for more than 5 years and attained a complete, deep, and durable molecular remission (molecular remission, 4.5; BCR-ABL ≤ .0032%) are the best candidates to consider stopping therapy.
2. In 50% of patients, a relapse with their disease will occur if the tyrosine kinase inhibitor is discontinued.
3. Almost all patients who progress by BCR-ABL RT-PCR quantitative testing can be successfully reinduced with the previous tyrosine kinase inhibitor.
However, the duration of remissions after a successful reinduction with a previous tyrosine kinase inhibitor or the depth of subsequent responses with reinduction of a previous tyrosine kinase inhibitor is not known. At this time, there are insufficient data to recommend routinely stopping tyrosine kinase inhibitors, even in this select group of patients.
Evidence (Imatinib mesylate) for AML
Several studies have suggested that the addition of imatinib to conventional combination chemotherapy induction regimens results in complete response rates, event-free survival rates, and OS rates that are higher than those in historical controls. At the present time, no conclusions can be drawn regarding the optimal imatinib dose or schedule.
1.In a study of imatinib combined with chemotherapy from the Northern Italy Leukemia Group, patients with newly diagnosed, untreated Ph1-positive ALL were treated with an induction regimen containing idarubicin, vincristine, prednisone, and L-asparaginase.
After accrual of an initial cohort, the study was modified to include the use of imatinib (600 mg qd from days 15 to 21).
In consolidation, patients received imatinib (600 mg qd for 7 days) beginning 3 days before the start of each course of chemotherapy.
• For all patients who achieved remission, the intent was to proceed to allogeneic transplant when and if an HLA-matched donor could be identified. Patients lacking a donor received an autologous transplant.
After completion of chemotherapy and transplant, all patients were to receive maintenance imatinib for as long as tolerated.
After 20 patients had accrued to the imatinib arm, L-asparaginase was omitted from the induction regimen from both arms because of toxicity.
• Outcomes for the first cohort of 35 patients (imatinib-free) were compared with those of the subsequent cohort of 59 (imatinib-treated) patients. For patients treated with imatinib, OS probability was 38% at 5 years (median, 3.1 y) versus 23% in the imatinib-free group (median, 1.1 y; P = .009).[Level of evidence: 3iii]
• The drawbacks of this nonrandomized study are the small sample size (94 total patients) and the change in the treatment regimen (omission of L-asparaginase) midway through the study. However, the results suggest that inclusion of imatinib into a relatively standard chemotherapy regimen for newly diagnosed adult patients with Ph1-positive ALL may provide a significant survival advantage.
2.In another study, ten patients with Ph1-positive ALL and ten patients with chronic myelogenous leukemia in lymphoid blast crisis were treated with doses of imatinib ranging from 300 mg to 1,000 mg per day. Of these 20 patients, four had complete hematologic remission and ten had marrow responses. Responses were short lived, with the majority of these patients relapsing at a median of 58 days after the start of therapy.
3.In another study, 48 patients with Ph1-positive ALL were treated with 400 mg to 800 mg of imatinib per day. The overall response rate was 60%, with 9 out of 48 patients (19%) achieving a complete remission. The responses again were short, with a median duration of 2.2 months.
In each of these studies, common toxicities were nausea and liver enzyme abnormalities, which necessitated interruption and/or dose reduction of imatinib. (Refer to the PDQ summary on Treatment-Related Nausea and Vomiting for more information.) Subsequent allogeneic transplant does not appear to be adversely affected by the addition of imatinib to the treatment regimen.
Imatinib is generally incorporated into the treatment of patients with Ph1-positive ALL because of the responses observed in monotherapy trials. If a suitable donor is available, allogeneic bone marrow transplantation should be considered because remissions are generally short with conventional ALL chemotherapy clinical trials
EGFR inhibitors
The epidermal growth factor receptor (EGFR/ erbB1/HER-1) belongs to a family of receptor tyrosine kinases that includes three other members (erbB2/HER-2, erbB3/HER-3, and erbB4/HER-4).
These receptors are anchored in the cytoplasmic membrane and share a similar structure that is composed of an extracellular ligand-binding domain, a short hydrophobic transmembrane region, and an intracytoplasmic tyrosine kinase domain.

EGF Receptor
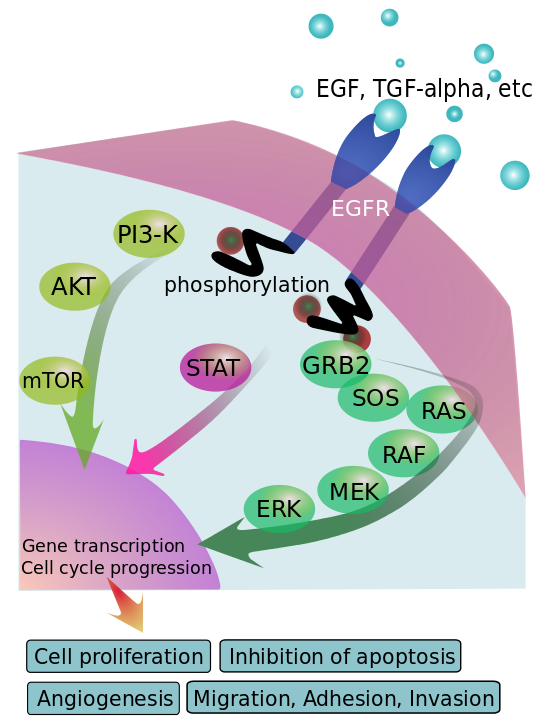
EGFR signaling pathway
EGFR is frequently expressed at abnormally high levels in epithelial tumors , and EGFR activation appears to be important in tumor growth and progression. Some types of cancers show mutations in their EGFRs, which may cause unregulated cell division through continual or abnormal activation of the EGFR.
EGFR inhibitors are drugs that bind to certain parts of the EGFR and inhibit cell growth.
There are two types of EGFR inhibitors:
1. Tyrosine kinase inhibitors (TKI) (eg, erlotinib, gefitinib) bind to the tyrosine kinase domain in the epidermal growth factor receptor and stop the activity of the EGFR.
erlotinib (Tarceva)
gefitinib (Iressa)
neratinib (Nerlynx)
lapatinib (Tykerb)
vandetanib (Caprelsa)
osimertinib (Tagrisso)
2. Monoclonal antibodies (eg, cetuximab, necitumumab) bind to the extracellular component of the EGFR and prevent epidermal growth factor from binding to the receptor, thus preventing cell activation and division.
cetuximab (Erbitux)
panitumumab (Vectibix)
necitumumab (Portrazza)
EGFR inhibitors may be used in the treatment of cancers that are caused by EGFR up-regulation, such as non-small-cell lung cancer, pancreatic cancer, breast cancer, and colon cancer.
erlotinib (Tarceva)
Oral epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI).
SUPPLIED As Oral Tab: 25mg, 100mg, 150mg
STORAGE: Store at 77 degrees F; excursions permitted to 59-86 degrees F
• Oral Administration: Erlotinib must be given orally on an empty stomach, 1 hour before or 2 hours after the ingestion of food. Administer at the same time each day.
• Coadministration of certain drugs may need to be avoided or dosage adjustments may be necessary; review drug interactions.
• Periodically monitor liver function, renal function, and electrolytes during treatment.
erlotinib monotherapy
■ For the treatment of metastatic non-small cell lung cancer (NSCLC) in patients after progression following at least one prior chemotherapy regimen
The tumors have epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations as detected by an FDA-approved test.
(Information on FDA-approved tests for the detection of EGFR mutations in NSCLC is available at: http://www.fda.gov/CompanionDiagnostics.)
As 1) first-line, 2) maintenance, or 3) second or greater line treatment after progression following at least one prior chemotherapy regimen.
erlotinib 150 mg PO once daily on an empty stomach (i.e., at least 1 hour before or 2 hours after food) until disease progression or unacceptable toxicity.
1) AS first-line thrapy In a randomized, multicenter, open label trial of patients with previously untreated metastatic NSCLC and EGFR exon 19 deletions or exon 21 (L858R) substitution mutations.
Despite a high crossover rate (82%), erlotinib monotherapy significantly improved progression-free survival (PFS) compared with platinum-based doublet chemotherapy(10.4 months vs. 5.2 months). Median overall survival (22.9 months vs. 19.5 months) and overall response rate (65% vs. 16%) were also improved.
2) In a separate trial, as maintenance therapy after first-line treatment with platinum-based chemotherapy for metastatic NSCLC,
erlotinib minimally but significantly improved median PFS (2.8 months vs. 2.6 months) and OS (12 months vs. 11 months) compared with placebo in a population that was 70% EGFR positive.
However, in a separate trial, erlotinib was not effective as maintenance therapy in patients without an EGFR exon 19 deletion or exon 21 (L858R) substitution mutation.
3) In a final study of patients with locally advanced or metastatic NSCLC after failure of at least one prior chemotherapy regimen,
OS was also significantly improved with erlotinib compared with placebo (6.7 months vs. 4.7 months).
Erlotinib was not effective in patients with locally advanced or metastatic NSCLC when administered concurrently with platinum-based chemotherapy.
erlotinib + gemcitabine
■ First-line treatment of locally advanced, unresectable or metastatic pancreatic cancer.
erlotinib 100 mg PO once daily on an empty stomach (i.e., at least one hour before or two hours after food)
in combination with gemcitabine 1,000 mg/m2 IV over 30 minutes once weekly for 7 consecutive weeks, followed by 1 week of rest. Beginning with week 9 (day 57), administer gemcitabine 1,000 mg/m2 IV over 30 minutes on days 1, 8, and 15, repeated every 28 days.
Continue treatment until disease progression or unacceptable toxicity occurs.
In a Phase 3 trial, the addition of erlotinib to gemcitabine significantly improved overall survival compared with gemcitabine plus placebo in patients with advanced pancreatic cancer.
erlotinib monotherapy
† For the treatment of recurrent or metastatic squamous cell head and neck cancer.
erlotinib 150 mg PO once daily has been studied.
Further study is needed to define the benefit of erlotinib in the treatment of head and neck cancer.
In a phase 2 trial, 115 patients treated with erlotinib had an overall response rate (ORR) of 4.3% with disease stabilization in 38.3% for a median duration of 16.1 months.
The median progression-free survival (PFS) was 9.6 weeks and the median overall survival (OS) was 6 months.
Subgroup analysis revealed a significant difference in overall survival favoring patients who developed at least grade 2 skin rashes compared to those who did not.
No difference in response was noted based upon HER1/EGFR expression.
MECHANISM OF ACTION
Erlotinib is a synthetic quinazolinamine that reversibly inhibits the kinase activity of the epidermal growth factor receptor (EGFR), preventing autophosphorylation of tyrosine residues associated with the receptor and thereby inhibiting further downstream signaling. EGFR is expressed on cell surfaces of both normal and cancer cells. In some tumor cells, signaling through this receptor plays a role in tumor cell survival and proliferation irrespective of EGFR mutation status. Erlotinib has a higher binding affinity for EGFR exon 19 deletion or exon 21 (L858R) substitution mutations compared to its affinity for the wild-type receptor; inhibition of other tyrosine kinase receptors by erlotinib has not been fully characterized.
DOSING CONSIDERATIONS
Hepatic Impairment
Baseline Hepatic Impairment: No dosage adjustment is necessary.
Treatment-Related Hepatotoxicity: Total bilirubin more than 3 times the upper limit of normal (ULN) or AST/ALT more than 5 times ULN in patients WITHOUT baseline hepatic impairment: Hold erlotinib therapy. Resume erlotinib at a reduced dose (by 50 mg decrements) when liver function tests resolve to baseline or less than or equal to grade 1. Discontinue erlotinib if resolution or significant improvement does not occur within 3 weeks.
Total bilirubin 2 times baseline or AST/ALT 3 times baseline in patients WITH pre-existing hepatic impairment, or biliary obstruction: Hold erlotinib therapy. Resume erlotinib at a reduced dose (by 50 mg decrements) when liver function tests resolve to baseline or less than or equal to grade 1. Discontinue erlotinib if resolution or significant improvement does not occur within 3 weeks.
Renal Impairment
Baseline Renal Impairment: No dosage adjustments are necessary.
Treatment-Induced Nephrotoxicity: Grade 3 or 4 renal impairment: Hold erlotinib therapy. When nephrotoxicity has resolved to baseline or less than or equal to grade 1, therapy may be resumed at a reduced dose (by 50 mg decrements). Alternatively, consider discontinuation of erlotinib.
Read the following informatin at PDR: erlotinib - Drug Summary
CONTRAINDICATIONS / PRECAUTIONS
ADVERSE REACTIONS
DRUG INTERACTIONS
PREGNANCY AND LACTATION
Note: Rash occurs in the majority of patients. This resembles acne and primarily involves the face and neck. It is self-limited and resolves in the majority of cases, even with continued use. Interestingly, some clinical studies have indicated a correlation between the severity of the skin reactions and increased survival though this has not been quantitatively assessed.[9] The Journal of Clinical Oncology reported in 2004 that "cutaneous [skin] rash seems to be a surrogate marker of clinical benefit, but this finding should be confirmed in ongoing and future studies."
gefitinib (Iressa)
Oral selective epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI).
Received accelerated approval for third-line treatment of advanced NSCLC (unselected for EGFR status) in 2003,
and was later withdrawn from the market after subsequent clinical trials failed to verify clinical benefit.
The current approval is for first-line treatment of EGFR mutation-positive metastatic NSCLC (exon 19 deletion or exon 21 (L858R) substitution mutation) after demonstrating an improvement in ORR in a multicenter, single-arm clinical trial.
An unplanned subgroup analysis of another clinical trial found an improvement in PFS and duration of response with gefitinib treatment over carboplatin/paclitaxel.
Serious adverse effects include interstitial lung disease, liver damage, gastrointestinal perforation, severe diarrhea and ocular disorders. The most common side effects are diarrhea and skin reactions.
SUPPLIED As Oral Tab: 250mg
STORAGE: Store at controlled room temperature (between 68 and 77 degrees F)
Standard gefitinib regimen
gefitinib 250 mg PO once daily (with or without food) until disease progression or unacceptable toxicity.
(In the absence of severe adverse drug reactions, increase the dose to 500 mg PO once daily in patients receiving concomitant strong CYP450 3A4 enzyme inducers.)
gefitinib monotherapy
■ For the first-line treatment of metastatic non-small cell lung cancer (NSCLC)
in patients whose tumors have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations as detected by an FDA-approved test.
(NOTE: Information on FDA-approved tests for the detection of EGFR mutations in NSCLC is available at http://www.fda.gov/CompanionDiagnostics.)
(NOTE: The safety and efficacy of gefitinib have not been established in patients with metastatic NSCLC whose tumors have EGFR mutations other than exon 19 deletions or exon 21 (L858R) substitution mutations.)
gefitinib 250 mg PO once daily until disease progression or unacceptable toxicity.
(In the absence of severe adverse drug reactions, increase the dose to 500 mg PO once daily in patients receiving concomitant strong CYP450 3A4 enzyme inducers.)
After a median treatment duration of 8 months, patients with EGFR mutation-positive metastatic NSCLC (n = 106) had an objective response rate (ORR) of 50% (95% CI, 41%—59%) with a median duration of response of 6 months (95% CI, 5.6—11.1 months) by blinded independent central review in a multicenter, single-arm, open-label clinical trial.
Response rates were similar in patients whose tumors had EGFR exon 19 deletions and exon 21 L858R substitution mutations.
An unplanned exploratory analysis of a randomized, multicenter, open-label clinical trial (n = 1217) found improved progression-free survival (PFS) (10.9 months vs. 7.4 months; HR 0.54; 95% CI, 0.38—0.79) and ORR (67% vs. 41%) in EGFR mutation-positive metastatic NSCLC treated with first-line gefitinib compared with carboplatin/paclitaxel.
gefitinib monotherapy
† For the treatment of recurrent or metastatic squamous-cell head and neck cancer.
gefitinib 250 or 500 mg/day PO has been studied; survival is not improved with gefitinib in this population.
In a randomized, phase III trial in 486 patients, median overall survival (OS) (primary endpoint) times were not improved with 250 mg/day PO (5.6 months; hazard ratio (HR) = 1.22; 95% CI, 0.95—1.57; p = 0.12) or 500 mg/day PO (6 months; HR = 1.12; 95% CI, 0.87—1.43; p = 0.39) compared with methotrexate 40 or 60 mg/m2 IV weekly (6.7 months).
Additionally, significantly worse OS was reported with 250 mg/day (HR = 1.62; 95% CI, 1.13—2.32; p = 0.01) or 500 mg/day (HR = 1.5; 95% CI, 1.06—2.13; p = 0.02) compared with methotrexate therapy in a subgroup of patients who had platinum-resistant disease.
Grade 3—5 tumor hemorrhage was reported in 4 patients (2.5%) who received gefitinib 250 mg/day and 2 patients (1.2%) who received gefitinib 500 mg/day.
gefitinib (Iressa)
Iressa is a tyrosine kinase inhibitor.
erlotinib monotherapy
■ For the first-line treatment of patients with metastatic non-small cell lung cancer (NSCLC)
The tumors have epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations as detected by an FDA-approved test.
Limitation of Use: Safety and efficacy of IRESSA have not been established in patients whose tumors have EGFR mutations other than exon 19 deletions or exon 21 (L858R) substitution mutations.
• The recommended dose of IRESSA is 250 mg orally once daily with or without food until disease progression or unacceptable toxicity.
(Do not take a missed dose within 12 hours of the next dose.)
• Administration to Patients Who Have Difficulty Swallowing Solids Immerse IRESSA tablets in 4 to 8 ounces of water by dropping the tablet in water, and stir for approximately 15 minutes. Immediately drink the liquid or administer through a naso-gastric tube. Rinse the container with 4 to 8 ounces of water and immediately drink or administer through the naso-gastric tube.
• CONTRAINDICATIONS: None.
• ADVERSE REACTIONS
The following adverse drug reactions are discussed in more detail in other sections of the labeling:
Interstitial Lung Disease
Hepatotoxicity
Gastrointestinal Perforation
Severe or Persistent Diarrhea
Ocular Disorders including Keratitis
Bullous and Exfoliative Skin Disorders
• Withhold IRESSA (for up to 14 days) for any of the following:
Acute onset or worsening of pulmonary symptoms (dyspnea, cough, fever)
NCI CTCAE Grade 2 or higher in ALT and/or AST elevations
NCI CTCAE Grade 3 or higher diarrhea
Signs and symptoms of severe or worsening ocular disorders including keratitis
NCI CTCAE Grade 3 or higher skin reactions
Resume treatment with IRESSA when the adverse reaction fully resolves or improves to NCI CTCAE Grade 1.
• Permanently discontinue IRESSA for:
Confirmed interstitial lung disease (ILD)
Severe hepatic impairment
Gastrointestinal perforation
Persistent ulcerative keratitis
• Dose Modifications for Drug Interactions
Strong CYP3A4 Inducers: Increase IRESSA to 500 mg daily in the absence of severe adverse drug reaction, and resume IRESSA at 250 mg seven days after discontinuation of the strong CYP3A4 inducer
The safety of IRESSA is based on the data from 2462 patients with NSCLC who received IRESSA 250 mg daily monotherapy in three randomized clinical studies (Study 2, Study 3 and Study 4).
Patients with a history of interstitial lung disease, drug-induced interstitial disease, radiation pneumonitis that required steroid treatment or any evidence of
clinically active interstitial lung disease were excluded from these studies.
Controlled Studies: Iressa monotherapy vs carboplatin/paclitaxel
Study 2 was a randomized, multicenter, open-label trial in which 1217 patients were randomized to receive first-line treatment for metastatic NSCLC;
607 patients received IRESSA 250 mg daily and 589 patients received carboplatin/paclitaxel.
The primary end point was progression-free survival.
The median duration of treatment with IRESSA was 5.9 months.
The study population characteristics were: median age 57 years, age less than 65 years (73%), female (79%), Asian (100%), NSCLC adenocarcinoma histology (100%), never smoker (94%), light ex-smoker (6%), ECOG PS 0 or 1 (90%).
Results:
a. The study met its primary objective of demonstrating the superiority of gefitinib compared with the carboplatin-paclitaxel combination for PFS (HR for progression or death, 0.74; 95% CI, 0.65–0.85; P < .001).
b. The median PFS was 5.7 months in the gefitinib group and 5.8 months in the carboplatin-paclitaxel group.[Level of evidence: 1iDiii]
c. Following the time that chemotherapy was discontinued and while gefitinib was continued, the PFS curves clearly separated and favored gefitinib.
■ The 12-month PFS rates were 24.9% with the gefitinib group and 6.7% with the carboplatin-paclitaxel group.
d. More than 90% of the patients in the trial with mutations had either del19 or exon 21 L858R mutations, which have been shown to be sensitive to EGFR inhibitors. In the subgroup of patients with a mutation, PFS was significantly longer among those who received gefitinib (HR, 0.48; 95% CI, 0.36–0.64; P < .001); however, in the subgroup of patients who were negative for a mutation, PFS was significantly longer in those who received the carboplatin-paclitaxel combination (HR with gefitinib, 2.85; 95% CI, 2.05–3.98; P < .001). There was a significant interaction between treatment and EGFR mutation with respect to PFS (P < .001).
e. OS was similar for patients who received gefitinib and carboplatin-paclitaxel, with no significant difference between treatments overall (HR, 0.90; 95% CI, 0.79–1.02; P = .109) or in EGFR mutation–positive (HR, 1.00; 95% CI, 0.76–1.33; P = .990) or EGFR mutation–negative (HR, 1.18; 95% CI, 0.86–1.63; P = .309; treatment by EGFR mutation interaction P = .480) subgroups. A high proportion (64.3%) of EGFR mutation–positive patients randomly assigned to the carboplatin-paclitaxel regimen received subsequent EGFR TKIs. PFS was significantly longer with gefitinib for patients whose tumors had both high EGFR gene copy number and EGFR mutation (HR, 0.48; 95% CI, 0.34–0.67) but significantly shorter when high EGFR gene copy number was not accompanied by EGFR mutation (HR, 3.85; 95% CI, 2.09–7.09).
† Indicates off-label use
cetuximab (Erbitux)
Monoclonal antibody that binds to and blocks the binding of ligands to the epidermal growth factor receptor.
FDA-approved for colorectal cancer and head and neck cancer.
SUPPLIED as Intravenous Inj Sol: 1mL, 2mg
• Can cause serious infusion reactions and cardiopulmonary arrest
■ For the treatment of KRAS wild-type metastatic colorectal cancer.
(NOTE: Cetuximab is not indicated for the treatment of RAS mutant colorectal cancer or when the results of RAS mutation tests are unknown. The absence of a RAS mutation should be confirmed by an FDA approved test prior to starting therapy.)
(NOTE: The serine-threonine kinase BRAF is the principal effector of KRAS. The introduction of the mutant BRAF V600E allele may impair therapeutic response to EGFR inhibitors. Mutant BRAF may be present in wild-type KRAS.)
cetuximab monotherapy
# For the treatment of KRAS wild-type, EGFR-expressing, metastatic colorectal cancer (mCRC) as a single agent after failure of both irinotecan- and oxaliplatin-based regimens or in patients who are intolerant to irinotecan-based chemotherapy.
cetuximab 400 mg/m2 IV over 120 minutes (maximum infusion rate: 10 mg/minute) on day 1,
followed by weekly infusions of 250 mg/m2 IV over 60 minutes (maximum infusion rate: 10 mg/minute) until disease progression or unacceptable toxicity.
In a multicenter, randomized, open-label clinical trial of patients with previously treated, recurrent mCRC, cetuximab monotherapy (n = 287) significantly improved median overall survival compared with best supportive care (n = 285) (6.1 months vs. 4.6 months; HR 0.77; p = 0.0046); in the subset of patients with KRAS wild-type tumors (n = 245), cetuximab monotherapy also improved overall survival (8.6 months vs. 5 months; HR 0.63).
cetuximab + irinotecan
# For the treatment of KRAS wild-type, EGFR-expressing, metastatic colorectal cancer (mCRC) in patients who are refractory to irinotecan-based chemotherapy.
(NOTE: In the clinical trials, patients were required to have immunohistochemical evidence of positive EGFR expression from primary tumor or metastatic tumor site using the DakCytomation EGFR pharmDx test kit. Response rate did not correlate with either the percentage of positive cells or the intensity of EGFR expression. EGFR expression status should be determined by an FDA approved test prior to starting therapy.)
cetuximab 400 mg/m2 IV over 120 minutes (maximum infusion rate: 10 mg/minute) on day 1,
followed by weekly infusions of cetuximab 250 mg/m2 IV over 60 minutes (maximum infusion rate: 10 mg/minute) until disease progression or unacceptable toxicity, in combination with irinotecan.
Multiple dosage regimens of irinotecan have been studied in combination with cetuximab, including irinotecan 180 mg/m2 IV once every 3 weeks; irinotecan 125 mg/m2 IV once weekly for 4 weeks out of 6; and, irinotecan 350 mg/m2 IV once every 3 weeks.
In a multicenter clinical trial of patients with recurrent mCRC (tumor specimens not available for KRAS testing), patients were treated with cetuximab alone (n = 111) or cetuximab plus irinotecan (n = 218); in the irinotecan arm, the irinotecan dose and schedule was the same as the patient had previously failed.
Of these patients, approximately 2/3 had previously failed oxaliplatin treatment.
The objective response rate was 23% for patients treated with cetuximab plus irinotecan, versus 11% for those who received cetuximab alone;
the median duration of response was 5.7 months vs. 4.2 months, and the median time to progression was 4.1 months versus 1.5 months, respectively.
Response rates to combination therapy and monotherapy were similar for irinotecan refractory patients and patients who had failed both irinotecan and oxaliplatin.
cetuximab (Erbitux)
■ For the treatment of head and neck cancer.
NOTE: Pretreatment assessment for evidence of EGFR expression is not required for patients with squamous cell cancer of the head and neck.
NOTE: Progression-free survival was not improved when cetuximab was added to radiation therapy and cisplatin for the treatment of locally advanced squamous cell carcinoma of the head and neck in a controlled study (n = 940); grade 3 and 4 toxicity and adverse reactions with fatal outcomes occurred more often in cetuximab-treated patients in this study.
cetuximab + Radiation
# For the treatment locally or regionally advanced squamous cell cancer of the head and neck.
cetuximab 400 mg/m2 IV over 2 hours (max rate: 5 mL/minute) as an initial loading dose 1 week prior to initiation of a course of radiation therapy.
The recommended maintenance dose is 250 mg/m2 IV over 60 minutes (max rate: 5 mL/minute) weekly for the duration of the radiation therapy (6 to 7 weeks).
Complete cetuximab administration 1 hour before radiation therapy.
In a clinical trial, patients with locoregionally advanced head and neck cancer were randomized to receive high-dose radiation therapy alone (n = 213) or high-dose radiation therapy plus cetuximab (n = 211).
The median duration of locoregional control was 24.4 months for patients treated with cetuximab plus radiation therapy vs. 14.9 months for patients treated with radiotherapy alone (hazard ratio for locoregional progression or death, 0.68; p = 0.005).
With a median follow-up of 54 months, the median duration of survival was 49 months among patients treated with combined therapy and 29.3 months for those treated with radiation therapy alone (hazard ratio for death 0.74; p = 0.03).
Cetuximab plus radiation therapy significantly prolonged progression-free survival (hazard ratio for disease progression or death 0.70; p = 0.006).
panitumumab (Vectibix)
Anti-EGFR monoclonal antibody
Used for wild-type RAS metastatic colorectal cancer as monotherapy or in combination with chemotherapy
Dermatologic adverse reactions and severe infusion reactions may occur
SUPPLIED as 20mg/ml Intravenous Sol: 100 mg/5 mL (20 mg/mL) and 400 mg/20 mL (20 mg/mL) in single-dose vials.
Store in original carton in refrigerator (35 to 46 degrees F) until time of use, Do not freeze
Protect from direct sunlight
Diluted product should be used within 6 hours if stored at room temperature or within 24 hours if stored at 36 to 46 degrees
Discard product if it contains particulate matter, is cloudy, or discolored
Discard unused portion. Do not store for later use
Intravenous Administration
Reconstitution:
Panitumumab solution should be colorless, but may contain a small amount of visible translucent-to-white, amorphous, proteinaceous particulates. Do not administer if there is any discoloration.
Using aseptic technique and a 21-gauge or larger needle, withdraw the necessary amount for a dose of 6 mg/kg. Do not use needle-free devices (e.g., vial adapters) to withdraw vial contents.
Vials of panitumumab do not contain preservatives; discard any unused portion remaining in the vial.
Preparation:
Dilute doses of 1,000 mg or less to a total volume of 100 mL with 0.9% sodium chloride injection.
Dilute doses higher than 1,000 mg to a total volume of 150 mL with 0.9% sodium chloride injection.
The final concentration should not exceed 10 mg/mL
.
Mix diluted solution by gentle inversion; do not shake.
Storage following dilution: The diluted infusion should be used within 6 hours of preparation if stored at room temperature, or within 24 hours if stored under refrigeration (2 to 8 degrees C or 36 to 46 degrees F); do not freeze.
Intravenous Infusion:
Administer only as an IV infusion via a controlled rate IV infusion pump or syringe pump using a low-protein binding 0.2 micron or 0.22 micron in-line filter; do not administer IV push or as a bolus injection.
Flush line with 0.9% sodium chloride injection before and after administration. Do not mix panitumumab with, or administer as an infusion with, other medicines or infusions.
Infuse doses of 1,000 mg or lower over 60 minutes through a peripheral line or indwelling catheter. If the first infusion is tolerated, subsequent doses of 1,000 mg or lower may be infused over 30 to 60 minutes. Infuse doses higher than 1,000 mg over 90 minutes.
Monitor for infusion-related reactions (e.g., fever, chills, dyspnea, bronchospasm, and hypotension).
• Infection, sepsis, serious rash, skin disease, sunlight (UV) exposure
Severe dermatologic and soft tissue toxicities have been commonly reported with panitumumab use.
Monitor patients who develop a serious rash for complications from inflammation or infection (e.g., necrotizing fasciitis, abscesses, and sepsis);
an interruption or discontinuation of therapy may be necessary for severe or life-threatening skin toxicity.
Subsequent dosage adjustment is required in patients who have therapy withheld due to dermatologic toxicity.
Advise patients receiving panitumumab to wear sunscreen, hats, and limit sunlight (UV) exposure during therapy and for 2 months after the last dose, as UV exposure can exacerbate any skin reactions that may occur.
Fatal or life-threatening bullous mucocutaneous skin disease with blisters, erosions, and skin sloughing have been observed;
it could not be determined whether these reactions were due to EGFR inhibition or idiosyncratic immune-related effects (e.g., Stevens-Johnson syndrome or toxic epidermal necrolysis).
The median time to development of dermatologic toxicity was 12 days after the first dose of panitumumab, with a median time to resolution of 98 days; dose interruption was required in 11% of patients.
Panitumumab should only be given in a hospital or clinic setting with full resuscitation equipment and under the supervision of a physician experienced with chemotherapy administration. Appropriate medical resources for the treatment of severe infusion reactions should be available. For individuals who experience infusion-related reactions, a prolonged infusion and observation period may be required.
panitumumab (Vectibix)
■ For the treatment of wild-type RAS (both KRAS and NRAS) metastatic colorectal cancer.
(Confirm the absence of a RAS mutation in exon 2 (codons 12 and 13), exon 3 (codons 59 and 61), and exon 4 (codons 117 and 146) of both KRAS and NRAS with an FDA-approved test prior to initiation of therapy.)
panitumumab + FOLFOX4 regimen
# For the first-line treatment of wild-type RAS (both KRAS and NRAS) metastatic colorectal cancer in combination with FOLFOX4.
panitumumab 6 mg/kg IV over 60 minutes (infuse doses higher than 1,000 mg over 90 minutes) on day 1, every 2 weeks prior to FOLFOX4 chemotherapy;
if the first infusion is tolerated, subsequent doses of 1,000 mg or lower may be infused over 30 to 60 minutes.
After completion of the panitumumab infusion, administer FOLFOX4 chemotherapy:
leucovorin 200 mg/m2 IV and oxaliplatin 85 mg/m2 IV (both over 120 minutes via Y-site) on day 1 followed by 5-fluorouracil (5-FU) 400 mg/m2 IV bolus. The 5-FU bolus should be followed by a continuous IV infusion of 5-FU 600 mg/m2 over 22 hours on day 1. On day 2, administer leucovorin 200 mg/m2 IV over 2 hours followed by 5-FU 400 mg/m2 IV bolus and 5-FU 600 mg/m2 IV continuous infusion over 22 hours.
The order of administration is panitumumab, followed by oxaliplatin and leucovorin, followed by 5-FU. This 2-day regimen is repeated every 2 weeks until disease progression or unacceptable toxicity.
In a multicenter, randomized, open-label trial of patients with previously untreated metastatic colorectal cancer,
panitumumab plus FOLFOX4 significantly improved progression-free survival (PFS) in the subgroups of patients with wild-type RAS (10.1 vs. 7.9 months) and wild-type KRAS (9.6 vs. 8 months) compared with FOLFOX4 alone;
the overall response rate was also improved in the panitumumab arm. Overall survival was estimated to be 23.3 months for combination therapy and 19.4 months with FOLFOX4 alone in patients with wild-type KRAS; results were similar in patients with wild-type RAS (25.8 vs. 20.2 months).
CONTRAINDICATIONS: None
WARNINGS AND PRECAUTIONS
Dermatologic and Soft Tissue Toxicity: Monitor for dermatologic and soft tissue toxicities and withhold or discontinue Vectibix for severe or
life-threatening complications. Limit sun exposure.
Increased tumor progression, increased mortality, or lack of benefit in patients with RAS-mutant mCRC.
Electrolyte Depletion/Monitoring: Monitor electrolytes and institute appropriate treatment.
Infusion Reactions: Terminate the infusion for severe infusion reactions.
Pulmonary Fibrosis/Interstitial Lung Disease (ILD): Permanently discontinue Vectibix in patients developing ILD.
Ocular Toxicities: Monitor for keratitis or ulcerative keratitis. Interrupt or discontinue Vectibix for acute or worsening keratitis.
Embryo-fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception during treatment with Vectibix and for 2 months after the last dose.
ADVERSE REACTIONS
Most common adverse reactions (≥ 20%) of Vectibix as monotherapy are skin rash with variable presentations, paronychia, fatigue, nausea, and diarrhea.
Most common adverse reactions (≥ 20%) of Vectibix as monotherapy are skin rash with variable presentations, paronychia, fatigue, nausea, and diarrhea.
Most common adverse reactions (≥ 20%) in clinical trials of Vectibix in combination with FOLFOX chemotherapy are diarrhea, stomatitis, mucosal
inflammation, asthenia, paronychia, anorexia, hypomagnesemia, hypokalemia, rash, acneiform dermatitis, pruritus, and dry skin.
† Indicates off-label use
Monoclonal Antibodies
trastuzumab (Herceptin)
Recombinant DNA-derived humanized IgG1 kappa monoclonal antibody against the HER2 protein
Used for breast and gastric cancers that overexpress HER2
Monitor left ventricular ejection fraction in all patients before, during, and after treatment; also watch for infusion-related reactions
Herceptin Intravenous Inj Pwd F/Sol: 150mg, 440mg
HER2-positive breast cancer
For the treatment of patients with HER2-positive breast cancer.
NOTE: Patients should be selected based on the presence of HER2 protein overexpression or HER2 gene amplification in tumor specimens. Information on FDA-approved tests for the detection of HER2 protein overexpression and HER2 gene amplification is available at http://www.fda.gov/CompanionDiagnostics. Tests should be specific for breast cancers due to differences in breast vs. gastric histopathology, including incomplete membrane staining and more frequent heterogeneous expression of HER2 seen in gastric cancers.
trastuzumab monotherapy
■ For the treatment of HER2-positive metastatic breast cancer in patients who have failed one or more chemotherapy regimens for metastatic disease as monotherapy.
trastuzumab 4 mg/kg IV over 90 minutes on day 1 (first week only), then 2 mg/kg IV over 30 minutes once weekly until disease progression.
Monotherapy with trastuzumab resulted in an overall response rate of 14% (complete response, 2%; partial response, 12%) in a multicenter, open-label, single-arm clinical trial in patients with relapse after 1 or more prior chemotherapy regimens for metastatic disease.
Complete responses only occurred in patients with disease limited to the skin and lymph nodes.
The response rate for patients with tumors that tested as CTA 3+ was 18%, while the response rate was 6% for tumors that tested as CTA 2+.
■ For adjuvant treatment of HER2-positive, node-positive or node-negative (ER/PR negative or with one high-risk feature) breast cancer as monotherapy, following multimodality anthracycline-based chemotherapy.
trastuzumab 8 mg/kg IV over 90 minutes on day 1 (of first cycle only), followed 3 weeks later by 6 mg/kg IV over 30 to 90 minutes, repeated every 21 days for a total of 52 weeks.
Trastuzumab therapy should begin within 3 weeks after completion of multi-modality, anthracycline-based chemotherapy.
Extending treatment beyond 1 year is not recommended as it increased the risk of asymptomatic cardiac dysfunction (2 years, 8.1%; 1 year, 4.6%) and grade 3 or higher adverse reactions (2 years, 20.4%; 1 year, 16.3%) without improving efficacy.
In a randomized clinical trial of patients with HER2-positive breast cancer, one year of adjuvant trastuzumab after the completion chemotherapy and radiation (if applicable) resulted in significantly improved disease free survival compared with observation; overall survival was not significantly improved.
trastuzumab + docetaxel
† For the treatment of HER2-positive metastatic breast cancer, in combination with docetaxel.
trastuzumab 4 mg/kg IV over 90 minutes on day 1 (of first cycle only), then 2 mg/kg IV over 30 minutes once weekly until disease progression or unacceptable toxicity,
in combination with docetaxel (100 mg/m2 IV on day 1) every 21 days for 6 to 8 cycles.
In a phase 2 trial of 186 patients with previously untreated HER2-positive metastatic breast cancer, docetaxel/trastuzumab significantly improved overall response rate (ORR) compared to docetaxel alone (61% vs 34%); time-to-progression (TTP) (11.7 vs. 6.1 months) and overall survival (OS) (31.2 vs. 22.7 months) were also significantly improved in the docetaxel/trastuzumab arm.
This benefit has been confirmed in other clinical trials, including a phase 3 trial of 263 patients where it was shown to produce a similar TTP and OS to docetaxel/carboplatin/trastuzumab, a regimen with known activity against breast cancer in the adjuvant setting.
In a phase 2 trial of 30 women with HER2-overexpressed metastatic breast cancer, docetaxel 35 mg/m2 IV on days 1, 8, and 15 was studied in combination with trastuzumab 2 mg/kg IV (4 mg/kg IV on day 0 of first cycle only) on days 1, 8, and 15, repeated every 28 days until disease progression or unacceptable toxicity; the ORR was 63%.
■ For adjuvant treatment of HER2-positive, node-positive or node-negative (ER/PR negative or with one high-risk feature) breast cancer in combination with docetaxel, after completion of doxorubicin and cyclophosphamide chemotherapy (AC-TH).
trastuzumab 4 mg/kg IV over 90 minutes on day 1 (of first cycle only), then 2 mg/kg IV over 30 minutes once weekly for a total of 12 weeks,
in combination with docetaxel (100 mg/m2 IV every 21 days) beginning on day 1 for a total of 4 cycles (12 weeks).
On week 13, begin trastuzumab 6 mg/kg IV over 30 to 90 minutes every 3 weeks as monotherapy for a total of 52 weeks of trastuzumab therapy.
Begin docetaxel plus trastuzumab after the completion of 4 cycles of AC chemotherapy (doxorubicin 60 mg/m2 IV and cyclophosphamide 600 mg/m2 IV every 21 days); trastuzumab should NOT be administered concurrently with doxorubicin and cyclophosphamide.
Extending treatment beyond 1 year is not recommended as it increased the risk of asymptomatic cardiac dysfunction (2 years, 8.1%; 1 year, 4.6%) and grade 3 or higher adverse reactions (2 years, 20.4%; 1 year, 16.3%) without improving efficacy.
In a randomized clinical trial of patients with HER2-positive breast cancer, adjuvant treatment with docetaxel plus trastuzumab after completion of AC chemotherapy significantly improved disease free survival compared with docetaxel alone.
trastuzumab + paclitaxel
■ For the first-line treatment of HER2-positive metastatic breast cancer, in combination with paclitaxel.
trastuzumab 4 mg/kg IV over 90 minutes on day 1 (of first cycle only), then 2 mg/kg IV over 30 minutes once weekly until disease progression, in combination with paclitaxel (175 mg/m2 IV over 3 hours on day 1) every 21 days for at least 6 cycles. In a multicenter, randomized, open-label clinical trial of patients with previously untreated metastatic breast cancer, treatment with trastuzumab plus paclitaxel significantly improved the median time to progression (6.7 vs. 2.5 months), overall response rate (38% vs. 15%), and duration of response (8.3 vs. 4.3 months) compared with paclitaxel alone; overall survival was not significantly improved.
trastuzumab + Chemotherapy
† For the treatment of HER2-positive metastatic breast cancer, in combination with vinorelbine.
REGIMEN 1 (every 3 weeks): 8 mg/kg IV over 90 minutes on day 1 (of first cycle only), followed 3 weeks later by 6 mg/kg IV over 30 minutes on day 1 every 3 weeks, in combination with vinorelbine (30 mg/m2 or 35 mg/m2 IV bolus on days 1 and 8) every 3 weeks, until disease progression or unacceptable toxicity. Administer vinorelbine after trastuzumab. In a phase 3 trial, patients with previously untreated locally advanced or metastatic breast cancer randomized to receive trastuzumab plus vinorelbine (n = 141) had a nonsignificant improvement in median time to progression (15.3 vs. 12.4 months) and overall survival (38.8 vs. 35.7 months) compared with trastuzumab plus docetaxel (n = 143). Grade 3 or 4 febrile neutropenia, leukopenia, infection, fever, neuropathy, and edema were significantly worse in the docetaxel arm. REGIMEN 2 (weekly): 4 mg/kg IV over 90 minutes on day 1 (of first cycle only), then 2 mg/kg IV over 30 minutes once weekly, in combination with vinorelbine (25 mg/m2 IV once weekly) beginning on day 1, until disease progression or unacceptable toxicity. Administer vinorelbine after trastuzumab. Trastuzumab plus vinorelbine had a response rate of 51% to 58% in two small clinical trials.
† For the treatment of HER2-positive, trastuzumab-refractory metastatic breast cancer, in combination with lapatinib.
Intravenous dosage
† For the treatment of HER2-positive, hormone receptor-positive metastatic breast cancer, in combination with anastrozole.
trastuzumab + Combination Chemotherapy
† For the front-line treatment of HER2-overexpressing metastatic breast cancer, in combination with carboplatin and paclitaxel.
REGIMEN 1: trastuzumab 4 mg/kg IV over 90 minutes on day 1 (of first cycle only), then 2 mg/kg IV over 30 minutes once weekly, in combination with paclitaxel (175 mg/m2 IV) followed by carboplatin (AUC 6 IV) administered on day 2 every 3 weeks, for at least 6 cycles. After chemotherapy is complete, continue trastuzumab 2 mg/kg IV over 30 minutes once weekly until disease progression or unacceptable toxicity. The primary endpoint of overall response rate was significantly increased with the addition of carboplatin to trastuzumab/paclitaxel compared with paclitaxel alone in a phase 3 clinical trial (52% vs. 36%). Progression-free survival was also superior in the carboplatin arm (10.7 vs. 7.1 months). Grade 4 neutropenia (36% vs. 12%) and grade 3 thrombocytopenia (9% vs. 1%) occurred more frequently in the carboplatin arm.
REGIMEN 2: trastuzumab 4 mg/kg IV over 90 minutes on day 1 (of first cycle only), then 2 mg/kg IV over 30 minutes once weekly, in combination with paclitaxel (80 mg/m2 IV over 1 hour) followed by carboplatin (AUC 2 IV over 15 minutes) on days 1, 8, and 15, repeated every 28 days for a maximum of 6 cycles. On days 1, 8, and 15 in the clinical trial, trastuzumab was administered immediately after paclitaxel and carboplatin infusions. After chemotherapy is complete, continue trastuzumab (2 mg/kg IV over 30 minutes once weekly, or 6 mg/kg IV every 3 weeks) until disease progression or unacceptable toxicity. In a parallel, multicenter phase II trial, weekly administration of paclitaxel/carboplatin/trastuzumab had an overall response rate of 81% compared to 65% with every-3-week therapy; median time to progression and median overall survival were also improved.
■ For adjuvant treatment of HER2-positive, node-positive or node negative (ER/PR negative or with one high-risk feature) breast cancer, in combination with carboplatin and docetaxel (TCH).
trastuzumab 4 mg/kg IV over 90 minutes on day 1 (of first cycle only), then 2 mg/kg IV over 30 minutes once weekly for a total of 18 weeks, in combination with docetaxel (75 mg/m2 IV) followed by carboplatin (AUC 6 IV over 30 to 60 minutes) every 3 weeks beginning on day 1 for a total of 6 cycles (18 weeks). On week 19, begin trastuzumab 6 mg/kg IV over 30 to 90 minutes every 3 weeks as monotherapy for a total of 52 weeks of trastuzumab therapy. Extending treatment beyond 1 year is not recommended as it increased the risk of asymptomatic cardiac dysfunction (2 years, 8.1%; 1 year, 4.6%) and grade 3 or higher adverse reactions (2 years, 20.4%; 1 year, 16.3%) without improving efficacy. In a randomized clinical trial, adjuvant treatment with trastuzumab in combination with docetaxel and carboplatin significantly improved disease free survival compared with doxorubicin plus cyclophosphamide (AC) followed by docetaxel.
† For the neoadjuvant treatment of HER2-positive breast cancer sequential to and in combination with doxorubicin, paclitaxel, and CMF chemotherapy.
trastuzumab 8 mg/kg IV over 90 minutes on day 1 (of cycle 1 only), followed 3 weeks later by 6 mg/kg IV over 30 minutes repeated every 3 weeks for 10 cycles, in combination with the following chemotherapy regimens. Doxorubicin (60 mg/m2 IV), followed by paclitaxel (150 mg/m2 IV over 3 hours) every 3 weeks for 3 cycles beginning on day 1 of trastuzumab therapy; then give paclitaxel (175 mg/m2 IV) alone every 3 weeks for an additional 4 cycles. After completion of doxorubicin/paclitaxel, administer cyclophosphamide (600 mg/m2 IV), methotrexate (40 mg/m2 IV), and 5-fluorouracil (600 mg/m2 IV) on days 1 and 8, every 4 weeks for 3 cycles (CMF); trastuzumab may be administered every 4 weeks during CMF chemotherapy. Patients with ER/PR-positive breast cancer should receive an antiestrogen agent per guidelines. Surgery followed by radiation therapy should be scheduled after completion of chemotherapy. In the clinical trial, additional cycles of trastuzumab were administered after surgery, beginning before or during radiotherapy, until the completion of one year total of trastuzumab therapy. In a phase 3 clinical trial, event-free survival was significantly improved at 3 years with the addition of trastuzumab to neoadjuvant doxorubicin/paclitaxel followed by CMF (71% vs. 56%) in patients with locally advanced HER2-positive breast cancer. In addition, the addition of trastuzumab significantly improved pathologic complete response rates compared to patients who did not receive trastuzumab (38% vs. 19%). Adverse events were similar between the treatment groups.
■ For adjuvant treatment of HER2-positive, node-positive or node negative (ER/PR negative or with one high-risk feature) breast cancer, in combination with carboplatin and docetaxel (TCH).
† For the treatment of HER2-positive, trastuzumab-resistant, advanced breast cancer in patients previously treated with a taxane, in combination with vinorelbine and everolimus.
trastuzumab 4 mg/kg IV over 90 minutes on day 1 (of first cycle only), then 2 mg/kg IV over 30 minutes once weekly, in combination with vinorelbine (25 mg/m2 IV once weekly) and everolimus (5 mg PO once daily) until disease progression or unacceptable toxicity. Coadministration of certain drugs may need to be avoided with everolimus or everolimus dosage adjustments may be necessary; review drug interactions. Everolimus plus vinorelbine and trastuzumab significantly improved progression-free survival compared with placebo plus vinorelbine and trastuzumab in patients with HER2-positive, trastuzumab-resistant advanced breast cancer in patients who had received prior taxane therapy; objective response rate and overall survival were not significantly improved.
trastuzumab + Pertuzumab + Chemotherapy
† For first-line treatment of HER2-positive metastatic breast cancer in combination with pertuzumab and docetaxel.
NOTE: Pertuzumab is FDA approved in combination with trastuzumab and docetaxel for the first-line treatment of HER2-positive metastatic breast cancer.
8 mg/kg IV over 90 minutes on day 1 (of first cycle only), followed 3 weeks later by trastuzumab 6 mg/kg IV over 30 to 90 minutes, repeated every 3 weeks until disease progression or unacceptable toxicity, in combination with pertuzumab (840 mg IV over 60 minutes initially, followed 3 weeks later by pertuzumab 420 mg IV over 30 to 60 minutes, repeated every 3 weeks until disease progression or unacceptable toxicity) and docetaxel (75 mg/m2 IV every 3 weeks for at least 6 cycles; dose may be escalated to 100 mg/m2 if initial dose is well tolerated). Pertuzumab and trastuzumab can be given in any order; however, both agents should precede docetaxel. If trastuzumab is given after pertuzumab, delay the infusion 30 to 60 minutes to observe for pertuzumab-related infusion reactions. Progression-free survival time was significantly improved in patients with HER2-positive metastatic breast cancer (CLEOPATRA study) treated with pertuzumab plus trastuzumab and docetaxel arm compared with those who received placebo plus trastuzumab and docetaxel (18.5 vs. 12.4 months) in a multicenter, double-blind, placebo-controlled trial. In a second interim analysis, median overall survival (OS) was also significantly improved in the pertuzumab-containing arm (not reached vs. 37.6 months).
BOXED WARNING
Angioedema, hypotension, infusion-related reactions, respiratory distress syndrome
Administration of trastuzumab products can result in infusion-related reactions characterized by fever and chills, nausea, vomiting, pain (in some cases at tumor sites), headache, dizziness, dyspnea, hypotension, rash, and asthenia. In postmarketing experience, serious and fatal infusion reactions have also been reported, including bronchospasm, anaphylaxis, angioedema, hypoxia, and severe hypotension. Infusion reactions were usually reported during or immediately following the initial infusion, but the onset and clinical course may vary, including progressive worsening, initial improvement followed by clinical deterioration, or delayed postinfusion events with rapid deterioration. For fatal events, death occurred within hours to days following a serious infusion reaction. Decrease the infusion rate for mild to moderate infusion reactions. Hold the infusion of trastuzumab product in all patients experiencing dyspnea or clinically significant hypotension, or if medical intervention (e.g., epinephrine, corticosteroids, diphenhydramine, bronchodilators, and oxygen) is required. Monitor patients until complete resolution of signs and symptoms. Discontinue trastuzumab product for anaphylaxis, angioedema, or acute respiratory distress syndrome; strongly consider discontinuation of therapy in patients with other severe reactions, as no data exist in regard to the most appropriate method of identification of patients who may safely be retreated with trastuzumab. Most patients who experienced a severe infusion reaction were premedicated with antihistamines and/or corticosteroids before resumption of trastuzumab infusion. Some patients tolerated trastuzumab, but some patients had recurrent severe infusion reactions despite premedications.
Asthma, chronic lung disease (CLD), chronic obstructive pulmonary disease (COPD), pneumonitis, pulmonary disease, pulmonary fibrosis, pulmonary toxicity
Use trastuzumab products with caution in patients with pre-existing chronic lung disease (CLD) such as chronic obstructive pulmonary disease (COPD), asthma, or pulmonary fibrosis. Trastuzumab products can cause serious and fatal pulmonary toxicity, including dyspnea, interstitial pneumonitis, pulmonary infiltrates, pleural effusions, non-cardiogenic pulmonary edema, pulmonary insufficiency and hypoxia, acute respiratory distress syndrome, and pulmonary fibrosis, which may be more severe in patients with symptomatic intrinsic lung disease or with more extensive tumor involvement of the lungs that results in dyspnea at rest. These reactions can also occur as sequelae of infusion reactions. Discontinue trastuzumab product for interstitial pneumonitis.
Cardiac arrhythmias, cardiac disease, cardiomyopathy, geriatric, heart failure, hypertension, ventricular dysfunction
Use trastuzumab products with caution in patients with a history of cardiac disease, heart failure, cardiomyopathy, ventricular dysfunction, hypertension, or cardiac arrhythmias, as trastuzumab products can cause these issues as well as cardiac death; an asymptomatic decline in the left ventricular ejection fraction (LVEF) is also possible. The risk of cardiac dysfunction was increased in geriatric patients compared to younger patients in clinical trials. Obtain a baseline measurement of LVEF by echocardiogram or MUGA immediately prior to initiation of trastuzumab products, every 3 months during treatment, upon completion of therapy, and every 6 months for at least 2 years after treatment for adjuvant therapy. An interruption or discontinuation of therapy may be necessary for a decline in LVEF; the safety of continuing or resuming trastuzumab product in patients with treatment-induced left ventricular cardiac dysfunction has not been adequately studied. However, 22 of 25 patients who developed left ventricular dysfunction during adjuvant trastuzumab therapy (after anthracycline treatment) had stable LVEF measurements and no CHF recurrence after rechallenge once symptoms were stable and patients were on maximum tolerated doses of ACE inhibitors and beta blockers. In another study, 16 of 26 patients with metastatic breast cancer who developed trastuzumab-related cardiotoxicity and fully recovered did not experience additional cardiotoxicity upon rechallenge; 10 patients had a subsequent cardiac event, with varying degrees of resolution after trastuzumab discontinuation. Trastuzumab was continued in 17 of 34 metastatic breast cancer patients with asymptomatic cardiac dysfunction in an additional retrospective study; 13 of these patients had complete recovery without specific cardiac treatment, 2 had complete recovery with treatment, and 2 were not assessed. Monitoring of the LVEF should occur more frequently, at 4 week intervals, if treatment is held for significant left ventricular cardiac dysfunction. Elevated troponin I immediately before or after trastuzumab administration may also be a predictor of cardiotoxicity. Patients who receive anthracycline therapy after stopping a trastuzumab product may be at increased risk of cardiac dysfunction; among patients receiving trastuzumab products as a single agent or in combination therapy, the highest absolute incidence of symptomatic cardiac dysfunction occurs when trastuzumab product is administered with an anthracycline.
Pregnancy
Fetal harm may occur if trastuzumab product is administered during pregnancy or within 7 months of conception; monitoring for oligohydramnios is recommended. If oligohydramnios occurs, fetal testing should be done that is appropriate for gestational age and is consistent with community standards of care. Pregnancy should be avoided by females taking trastuzumab product during therapy and for 7 months after the last dose; women who become pregnant during this time frame should be apprised of the potential hazard to the fetus. Encourage these women to enroll in the MotHER Pregnancy Registry by contacting 1-800-690-6720 or visiting http://www.motherpregnancyregistry.com. Healthcare professionals and patients should additionally report the pregnancy to the pregnancy pharmacovigilance program at Genentech (1-888-835-2555). Postmarketing, oligohydramnios and oligohydramnios sequence (manifesting as pulmonary hypoplasia, skeletal abnormalities, and neonatal death) occurred in pregnant women who received trastuzumab alone or in combination with chemotherapy. In some women, the amniotic fluid index increased after trastuzumab cessation. In one case, treatment was resumed with trastuzumab after the amniotic fluid index improved, resulting in a recurrence of oligohydramnios.
Contraception requirements, pregnancy testing, reproductive risk
Counsel patients about the reproductive risk and contraception requirements during treatment with trastuzumab product. Trastuzumab can be teratogenic if taken by the mother during pregnancy or within 7 months prior to conception. Females should avoid pregnancy and use effective contraception during and for at least 7 months after treatment with trastuzumab product. Females of reproductive potential should undergo pregnancy testing prior to initiation of trastuzumab product. Women who become pregnant while receiving trastuzumab or within 7 months of the last dose should be apprised of the potential hazard to the fetus and encouraged to enroll in the MotHER Pregnancy Registry (1-800-690-6720 or www.motherpregnancyregistry.com); additionally, healthcare providers and patients should report trastuzumab exposure to the pregnancy pharmacovigilance program at Genentech (1-888-835-2555).
HER2-positive metastatic gastric cancer or gastroesophageal junction adenocarcinoma
■ For the treatment of patients with previously untreated HER2-positive metastatic gastric cancer or gastroesophageal junction adenocarcinoma, in combination with cisplatin and 5-fluorouracil or capecitabine.
NOTE: Patients should be selected based on the presence of HER2 protein overexpression or HER2 gene amplification in tumor specimens. Information on FDA-approved tests for the detection of HER2 protein overexpression and HER2 gene amplification is available at http://www.fda.gov/CompanionDiagnostics. Tests should be specific for gastric cancers due to differences in breast vs. gastric histopathology, including incomplete membrane staining and more frequent heterogeneous expression of HER2 seen in gastric cancers.
trastuzumab 8 mg/kg IV over 90 minutes on day 1 (of first cycle only), followed 3 weeks later by 6 mg/kg IV over 30 to 90 minutes every 21 days until disease progression or unacceptable toxicity,
in combination with cisplatin (80 mg/m2 IV over 2 hours on day 1) plus either 5-fluorouracil (800 mg/m2/day by continuous IV infusion on days 1, 2, 3, 4, and 5) or capecitabine (1,000 mg/m2 PO twice daily on days 1 through 14), every 21 days for 6 cycles.
The primary endpoint of median overall survival was significantly improved in previously untreated patients with metastatic gastric cancer who received trastuzumab plus cisplatin and 5-FU or capecitabine (n = 298) compared with chemotherapy alone (n = 296) (13.1 months vs. 11.7 months) in open-label clinical trial.
† Indicates off-label use
rituximab (Rituxan)
Chimeric monoclonal anti-CD20 antibody
Used for rheumatoid arthritis, non-Hodgkin's lymphoma (NHL), chronic lymphocytic leukemia (CLL), and other autoimmune and malignant conditions
Rituxan Intravenous Inj Sol: 1mL, 10mg
Non-Hodgkin's lymphoma (NHL).
rituximab monotherapy
■ For patients with relapsed or refractory low-grade or follicular, CD20 positive, B-cell NHL.
rituximab 375 mg/m2 IV once weekly for 4 doses
produced an overall response rate (ORR) of 48%.
Response rates are higher in patients with International Working Formulation (IWF) B, C, and D histologic subtypes as compared to IWF A subtypes (58% vs. 12%).
Patients with smaller lesions (i.e. < 5 cm) and those previously treated with autologous bone marrow transplant have response rates of 55% and 78%, respectively.
Patients who progress after the initial 4 doses may be retreated with 4 additional doses of 375 mg/m2 IV once weekly.
This repeated schedule produced an ORR of 57% with a median duration of response of 13.4 months.
There is limited data regarding more than 2 courses.
In patients with bulky disease (single lesion > 10 cm), who were excluded from the previous study, rituximab 375 mg/m2 IV once weekly for 4 doses resulted in an ORR of 36% (3% complete response, 33% partial response) with a median response duration of 6.9 months. These patients had a higher incidence of Grade 3 and 4 adverse reactions.
■ As single-agent maintenance therapy in patients with low-grade, CD20-positive, B-cell non-Hodgkin's lymphoma (NHL) with nonprogressing disease (stable disease or better) following first-line treatment with cyclophosphamide, vincristine, and prednisone (CVP).
rituximab 375 mg/m2 IV weekly for 4 weeks repeated every 6 months for 2 years (total of 16 doses) as maintenance therapy starting 4 weeks after the completion of first-line chemotherapy with 6 to 8 cycles of cyclophosphamide, vincristine, and prednisone' (CVP)
This regimen was evaluated in previously untreated patients with advanced indolent lymphoma (including follicular lymphoma (FL)) in a randomized, phase III study.
Patients who responded or had stable disease to CVP induction therapy received either rituximab maintenance (n=158; FL, n=115) or observation (n=153; FL, n=113).
At a median follow-up time of 3.7 years,
the median progression-free survival (PFS) time (primary endpoint) was 4.3 years in the rituximab arm compared with 1.3 years in the observation arm (hazard ratio (HR) = 0.4; 95% CI, 0.3—0.5; p = 4.4 x 10-10).
The 3-year PFS rate (68% vs 33%; p < 0.001) but not the 3-year overall survival rate (92% vs 86%; HR = 0.6; 95% CI, 0.4—1.1) was significantly improved with rituximab maintenance.
rituximab + Chemothrapy
■ For first-line treatment of follicular, CD20-positive, B-cell NHL, in combination with chemotherapy.
rituximab 375 mg/m2 IV on day 1 of each cycle of chemotherapy for up to 8 infusions.
Rituximab has been studied in combination with first-line therapy consisting of
cyclophosphamide 750 mg/m2 IV on day 1, vincristine 1.4 mg/m2 (Max of 2 mg) IV on day 1, and prednisone 40 mg/m2/day PO on days 1—5 (R-CVP);
cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP);
cyclophosphamide, doxorubicin, etoposide, prednisone, and interferon alfa-2a (R-CHVP+I); and
mitoxantrone, chlorambucil, and prednisolone (R-MCP) in randomized, clinical trials.
R-CVP was compared with CVP (repeated every 21 days for up to 8 cycles) in 361 previously untreated patients with stage III or IV follicular lymphoma in a randomized, phase III study.
In a preplanned interim analysis (after 189 events), time to progression (TTP) was significantly improved in patients who received R-CVP compared with CVP alone and this study was terminated. At a median follow-up time of 53 months, the median TTP was 34 months in the R-CVP arm compared with 15 months in the CVP arm (p < 0.0001). Additionally, the 4-year overall survival rates were significantly improved with R-CVP (83% vs 77%; p = 0.029). Grade 3 or 4 neutropenia occurred more often with R-CVP compared with CVP (24% vs 14%).
■ As single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response following first-line treatment with rituximab in combination with chemotherapy.
rituximab 375 mg/m2 IV every 8 weeks for 12 doses as maintenance therapy starting 8 weeks after the completion of induction chemotherapy
The induction chemotherapy was with 8 doses of rituximab plus 6—8 cycles of cyclophosphamide, vincristine, and prednisone (R-CVP); 4—6 cycles of cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP); or 4—6 cycles of fludarabine, cyclophosphamide, and mitoxantrone (R-FCM)
The regimen was evaluated in previously untreated patients with follicular lymphoma in a multinational, randomized study.
Following a complete or partial response to induction therapy, patients (age range, 22—84 years) received either rituximab maintenance (n=505) or observation (n=514).
In a preplanned interim analysis (after 267 events), progression-free survival (PFS) was significantly improved with rituximab maintenance compared with observation only and this study was terminated. At a median follow-up time of 36 months, the 3-year PFS rate was 74.9% in the rituximab arm and 57.6% in the observation arm (p < 0.0001), and the risk of progression was significantly reduced with rituximab maintenance (hazard ratio = 0.55; 95% CI, 0.44—0.68). Overall survival was not significantly different between the 2 study arms with few deaths reported in either arm. Grade 2-4 infections were reported significantly more often with rituximab maintenance (39% vs 24%; p < 0.0001).
rituximab + ibritumomab tiuxetan
■ For the treatment of follicular, CD20-positive NHL, in combination with ibritumomab tiuxetan.
According to the manufacturer, rituximab 250 mg/m2 IV should be administered within 4 hours prior to the administration of indium (In)-111-ibritumomab followed 7—9 days later by rituximab 250 mg/m2 IV given within 4 hours prior to the administration of yttrium (Y)-90-ibritumomab.
However, in November 2011, the US Food and Drug Administration removed the requirement for imaging with In-111-ibritumomab.
The Y-90-ibritumomab therapeutic regimen consists of 2 rituximab infusions given on day 1 and on day 7, 8, or 9 plus one Y-90-ibritumomab dose given within 4 hours after the second rituximab infusion.
The dose for each rituximab infusion is 250 mg/m2 IV (initial rate of 50 mg/hour, increased in 50 mg/hour increments every 30 minutes to a maximum of 400 mg/hour).
Premedicate patients with diphenhydramine 50 mg PO and acetaminophen 650 mg PO 30 minutes prior to each rituximab dose.
Decrease the rate of rituximab infusion by 50% for mild to moderate infusion reactions;
the infusion may be restarted at 50% of the previous infusion rate when the reaction completely resolves.
Immediately stop the rituximab infusion for serious infusion reactions and discontinue the Y-90-ibritumomab therapeutic regimen.
In patients with previously untreated follicular NHL who achieved a partial or complete response to first-line chemotherapy, the dose of Y-90-ibritumomab is 0.4 millicuries (mCi)/kg (14.8 megabecquerels (MBq)/kg) IV over 10 minutes.
In patients with relapsed or refractory low-grade or follicular NHL who have a normal platelet count (>= 150,000 cells/mm3), the dose of Y-90-ibritumomab is 0.4 mCi/kg IV over 10 minutes.
In patients with relapsed or refractory low-grade or follicular NHL who have a platelet count of >= 100,000 to <= 149,000 cells/mm3, the dose of Y-90-ibritumomab is 0.3 mCi/kg (11.1 MBq/kg) IV over 10 minutes.
Y-90-ibritumomab should not be administered in patients with relapsed or refractory low-grade or follicular NHL who have a platelet count < 100,000 cells/mm3.
Use the patient’s actual body weight to calculate the Y-90-ibritumomab dose; do not exceed 32 mCi (1184 MBq).
Immediately stop the infusion and restart in another limb if signs or symptoms of extravasation occur.
rituximab + Chemotherapy
■ For first-line treatment of diffuse large B-cell, CD20-positive non-Hodgkin's lymphoma (NHL), in combination with CHOP or other anthracycline-based chemotherapy regimen.
Adults 18 to 59 years
rituximab 375 mg/m2 IV on day 1 of each cycle of chemotherapy for up to 8 infusions.
Rituximab has been studied in combination with 6 cycles of cyclophosphamide 750 mg/m2 IV on day 1, doxorubicin 50 mg/m2 IV on day 1, vincristine 2 mg IV on day 1, and prednisone 100 mg/day PO on days 1—5 repeated every 21 days (CHOP-21) or CHOP-like chemotherapy in 824 adult patients less than 60 years of age with previously untreated, good prognosis, diffuse large B-cell lymphoma in a large, multinational, randomized, clinical trial.
CHOP-like chemotherapy consisted of CHOP-21 plus etoposide (CHOEP-21); methotrexate, doxorubicin, cyclophosphamide, vincristine, prednisone, and bleomycin (MACOP-B); or
prednisone, mitoxantrone, cyclophosphamide, etoposide, bleomycin, and vincristine (PMitCEBO).
In a preplanned interim analysis (after 100 events), event-free survival (EFS) was significantly improved with rituximab plus CHOP-like chemotherapy compared with CHOP-like chemotherapy alone and this study was terminated.
At a median follow-up of 72 months, the 6-year EFS (74.3% vs 55.8%; hazard ratio = 0.4; 95% CI, 0.32—0.62; p < 0.0001), progression-free survival (80.2% vs 63.9%; p < 0.0001), and overall survival (90.1% vs 80%; p = 0.0004) rates were significantly higher with rituximab plus CHOP-like chemotherapy compared with CHOP-like chemotherapy alone.
Adults and Elderly patients >= 60 years
rituximab 375 mg/m2 IV on day 1 of each cycle of chemotherapy for up to 8 infusions.
Rituximab has been studied in combination with cyclophosphamide 750 mg/m2 IV on day 1, doxorubicin mg/m2 IV on day 1, vincristine 1.4 mg/m2 (Max of 2 mg) IV on day 1, and prednisone 40 or 100 mg/m2/day PO on days 1—5 (R-CHOP) repeated every 21 days for up to 8 cycles in patients >= 60 years of age with previously untreated diffuse large B-cell lymphoma in randomized, clinical trials.
In a randomized, phase III study in 632 patients aged 60 years or older (range, 60—92 years), the 3-year failure-free survival rate was significantly higher with R-CHOP compared with CHOP (53% vs 46%; hazard ratio (HR) = 0.78; 95% CI, 0.61—0.99; p = 0.04) at a median follow-up of 3.5 years.
However, overall survival (OS) was not significantly improved in the R-CHOP arm (HR = 0.83; 95% CI, 0.63—1.09).
In another randomized trial, the median progression-free survival (PFS) (4.8 vs 1.2 years; p < 0.0001) and OS (8.4 vs 3.5 years; p < 0.0001) times were significantly improved with R-CHOP compared with CHOP in 399 patients aged 60 to 75 years.
The 10-year PFS rates were 36.5% and 20.1% in the R-CHOP and CHOP arms, respectively, and the 10-year OS rates were 43.5% and 27.6%, respectively.
rituximab + Chemotherapy
† For the treatment of relapsed or refractory, aggressive, CD20-positive NHL in transplant eligible patients, in combination with gemcitabine, dexamethasone, and cisplatin.
rituximab 375 mg/m2 IV on day 1 in combination with gemcitabine 1,000 mg/m2 IV on days 1 and 8; dexamethasone 40 mg orally daily on days 1, 2, 3, and 4; and cisplatin 75 mg/m2 IV on day 1 (R-GDP regimen) repeated every 21 days for 2 cycles was evaluated in a randomized, phase III trial (NCIC-CTG LY.12 trial).
Patients in the trial could receive a third cycle of therapy if they did not achieve a complete or partial response after the second cycle. Patients with CD20-positive lymphoma who received an autologous stem-cell transplant (ASCT) were randomized to receive either rituximab 375 mg/m2 IV every 2 months for 6 cycles or observation starting 28 days post ASCT.
† For the treatment of previously treated indolent NHL (including follicular lymphoma), in combination with bendamustine.
rituximab 375 mg/m2 IV on day 1 in combination with bendamustine (90 mg/m2 IV on days 2 and 3) every 28 days for 4—6 cycles.
In a phase II trial of 67 patients with relapsed indolent B-cell NHL or mantle-cell lymphoma, patients received B-R.
An additional dose of rituximab was administered 1 week prior to the first cycle of B-R and 4 weeks after the last cycle.
Patients receiving B-R had an overall response rate of 92% with a median progression-free survival of 23 months.
Grade 3 or 4 neutropenia (36%) and thrombocytopenia (9%) occurred. Several dose adjustment criteria were used in the study. For example, in the event of grade 3 nonhematologic or grade 4 hematologic toxicity, the bendamustine dose was reduced to 60 mg/m2 in the subsequent cycle.
If a similar severity toxicity occurred at the reduced dose, bendamustine was discontinued.
In another phase II trial, 57 of 63 patients (ORR 90%) with relapsed or refractory mantle-cell or low-grade lymphomas responded to B-R and median PFS was 24 months.
† For the treatment of previously untreated indolent NHL (including follicular lymphoma), in combination with bendamustine.
rituximab 375 mg/m2 IV on day 1 in combination with bendamustine (90 mg/m2 IV on days 1 and 2) repeated every 28 days (B-R regimen) for up to 6 cycles
This regimen has been evaluated in a phase III trial that compared B-R to standard R-CHOP in 514 patients with indolent or mantle cell lymphomas.
† For the treatment of follicular NHL, in combination with CHOP chemotherapy.
† For the treatment of previously untreated follicular NHL, in combination with fludarabine and mitoxantrone.
† For the first-line treatment of indolent NHL (including follicular lymphoma), in combination with lenalidomide.
Mantle cell lymphoma
† For the treatment of previously untreated advanced mantle cell lymphoma (MCL), in combination with CHOP chemotherapy.
† For the treatment of previously untreated advanced mantle cell lymphoma (MCL), in combination with hyper-CVAD chemotherapy.
† For the treatment of previously untreated mantle cell lymphoma (MCL), in combination with bendamustine.
† For the treatment of previously treated mantle cell lymphoma (MCL), in combination with bendamustine.
† For the treatment of mantle cell lymphoma (MCL), in combination with lenalidomide.
† As maintenance therapy.
BOXED WARNING
Serious rash
Mucocutaneous reactions including serious rash such as paraneoplastic pemphigus, Stevens-Johnson syndrome, lichenoid dermatitis, vesiculobullous dermatitis, and toxic epidermal necrolysis have been reported with rituximab use; some cases were fatal. The time to onset of mucocutaneous reactions has varied; however, there have been reports of reactions occurring with the first rituximab dose. Discontinue rituximab in patients who develop a severe mucocutaneous reaction; the safety of restarting rituximab in these patients has not been evaluated.
Hepatitis B exacerbation
Hepatitis B virus (HBV) reactivation resulting in hepatitis B exacerbation, fulminant hepatitis, hepatic failure, and death may occur during or following treatment with an anti-CD20 antibody, including rituximab. Screen all patients for HBV (e.g., HBsAg and anti-HBc titers) prior to starting rituximab therapy. In patients who have evidence of HBV, consult with an expert in hepatitis management for monitoring and treatment recommendations. Additionally, monitor all patients with current or prior HBV for signs of hepatitis or HBV reactivation during and for several months after therapy. In patients who develop HBV reactivation, discontinue rituximab and any concomitant chemotherapy and initiate appropriate treatment. The safety of restarting rituximab in these patients has not been evaluated.
Immunosuppression, progressive multifocal leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML), caused by the JC virus, has been reported in patients with hematologic malignancies and autoimmune diseases who received rituximab; some cases were fatal. Evaluate patients who develop new neurologic symptoms; consider consulting a neurologist and obtaining a brain MRI and lumbar puncture. Discontinue rituximab in patients who develop PML; additionally, consider discontinuing or reducing concomitant chemotherapy or immunosuppressive therapy. Most patients with hematologic malignancies who developed PML received rituximab in combination with chemotherapy or as part of a hematopoietic stem-cell transplant; patients with autoimmune diseases who developed PML had received prior or concurrent immunosuppression therapy. Most PML cases occurred within 12 months of the final rituximab infusion.
Infusion-related reactions, pulmonary disease
Infusion-related reactions have been reported with rituximab use; some cases were fatal. Premedicate with acetaminophen and an antihistamine prior to each rituximab infusion; methylprednisolone administration is also recommended prior to each infusion in patients with rheumatoid arthritis and pemphigus vulgaris. Interrupt or permanently discontinue the rituximab infusion in patients who develop infusion reactions; an infusion rate reduction is recommended if the infusion is resumed. Closely monitor patients with preexisting cardiac disease or pulmonary disease, patients with a prior history of cardiopulmonary adverse reactions, and patients with high numbers of circulating malignant cells (i.e., 25,000 cells/mm3 or more). Signs and symptoms or sequelae of infusion reactions include anaphylaxis, angioedema, bronchospasm, cardiogenic shock, flushing, hypotension, hypoxia, myocardial infarction, pulmonary infiltrates, throat irritation, tremor, urticaria, and ventricular fibrillation; acute respiratory distress syndrome and cytokine release syndrome have also been reported.
Chronic lymphocytic leukemia (CLL).
rituximab + Chemotherapy
■ For the treatment of CD20-positive CLL, in combination with cyclophosphamide and fludarabine.
NOTE: Pneumocystis jirovecii pneumonia and antiviral prophylaxis is recommended for patients with chronic lymphocytic leukemia during treatment and for up to 12 months after treatment as appropriate.
rituximab 375 mg/m2 IV on cycle 1; give the day prior to (day 0) fludarabine and cyclophosphamide administration.
On cycles 2 to 6, give rituximab 500 mg/m2 IV on day 1 in combination with fludarabine and cyclophosphamide; treatment cycles are repeated every 28 days.
Rituximab plus fludarabine 25 mg/m2/day on days 1, 2, and 3 and cyclophosphamide 250 mg/m2/day on days 1, 2, and 3 repeated every 28 days (FCR) for 6 cycles has been studied in randomized, phase III trials.
The progression-free survival (PFS) time (primary endpoint) was significantly improved with FCR (mean of 5.2 cycles) compared with fludarabine and cyclophosphamide (FC) alone (51.8 months vs. 32.8 months; p < 0.0001) in previously untreated CLL patients in a multinational, randomized, phase III trial (CLL8 trial; n = 817).
At a median follow-up time of 3.1 years, the 3-year PFS (65% vs. 45%; hazard ratio (HR) = 0.56; 95% CI, 0.46 to 0.69) and overall survival (OS) (87% vs. 83%; HR = 0.67; 95% CI, 0.48 to 0.92) rates were also significantly improved with FCR.
At a median follow-up of 5.9 years, the median PFS times were 56.8 months and 32.9 months in the FCR and FC arms, respectively (HR = 0.59; 95% CI, 0.5 to 0.69); the 5-year PFS rates were 46.8% and 25.5%, respectively.
Additionally, the median OS times were not yet reached and 86 months in the FCR and FC arms, respectively (HR = 0.68; 95% CI, 0.54 to 0.89); the 5-year OS rates were 78.7% and 66.9%, respectively. The median PFS time (primary endpoint) was 30.6 months with FCR compared with 20.6 months with FC (HR = 0.65; 95% CI, 0.51 to 0.82; p < 0.001) in CLL patients who had relapsed or refractory disease following 1 prior line of therapy in another multinational, randomized, phase III trial (n = 552).
All patients in this study received tumor lysis and antibiotic/antiviral prophylaxis.
At a median follow-up time of 25 months, the median OS was not significantly different between treatment arms (FCR, median time not reached; FC, 52 months). There were more treatment-related deaths reported with R-FC therapy (19 vs. 14 deaths).
† For the first-line treatment of CLL, in combination with fludarabine.
rituximab 375 mg/m2 IV for 4 weekly doses in patients with stable disease or better after 2 months of observation following six 28-day cycles of fludarabine 25 mg/m2 per day IV on days 1 to 5 (sequential therapy) OR
rituximab 375 mg/m2 IV on day 1 and 4 on cycle 1 then 375 mg/m2 IV on day 1 only on cycles 2 to 6 concurrently with six 28-day cycles of fludarabine 25 mg/m2 per day IV on days 1 to 5 followed by rituximab 375 mg/m2 IV for 4 weekly doses in patients who had stable disease or better 2 months after therapy (concurrent therapy) was studied in 104 patients in a randomized, noncomparative, phase II trial.
Because the first 44 patients treated with concurrent therapy experienced infusion-related adverse effects, the last 7 patients received stepped up therapy on cycle 1 with rituximab 50 mg/m2 IV over 4 hours on day 1, 325 mg/m2 IV starting at 50 mg/hr titrated to a maximum rate of 400 mg/hr on day 3, and 375 mg/m2 IV at 100 mg/hr for the first 15 minutes with rate increases to complete the infusion in the next 45 minutes on day 5.
All patients received allopurinol 300 mg/day for the first 14 days of treatment.
At 2 months following the completion of therapy, the overall response rates (ORR) were 77% (complete response (CR) rate, 28%) and 90% (CR rate, 47%) with sequential and concurrent therapy, respectively.
The 2-year progression-free survival (PFS) rate was 70% with both sequential and concurrent therapy at a median follow-up of 23 months.
In the overall study population, the median PFS and overall survival (OS) times were 42 and 85 months, respectively, and the estimated 5-year PFS and OS rates were 28% and 71%, respectively, at a median follow-up of 117 months.
Grade 3 and 4 hematologic toxicity was more common with concurrent therapy; however, infection rates were similar with both therapies and no therapy-related myeloid neoplasms were reported at long-term follow-up.
In a retrospective, comparative analysis of the 104 patients from this study (CALGB 9712) and the fludarabine only arm (n=178) from another randomized study (CALGB 9011) in previously untreated CLL patients, combination therapy with rituximab plus fludarabine was associated with a significantly improved CR rate (38% vs. 20%; p = 0.002), ORR (84% vs 63%; p = 0.0003), PFS (p < 0.0001), and OS (p = 0.003) compared with fludarabine alone.
† For the first-line treatment of CLL in elderly patients, in combination with chlorambucil.
† For the treatment of CLL, in combination with cyclophosphamide and pentostatin.
† For the treatment of relapsed or refractory CLL, in combination with idelalisib and bendamustine.
† For the treatment of CLL in patients who have received at least 1 prior therapy, in combination with venetoclax.
NOTE: Venetoclax is FDA approved in combination with rituximab for the treatment of CLL or SLL in patients with or without a 17p deletion who have received at least 1 prior therapy.
Rheumatoid arthritis
rituximab + Methotrexate
■ For reducing signs and symptoms of moderately- to severely-active rheumatoid arthritis in combination with methotrexate in patients who have had an inadequate response to one or more tumor necrosis factor (TNF) antagonist therapies.
NOTE: In patients with rheumatoid arthritis, obtain a CBC at 2—4 month intervals during rituximab therapy.
rituximab 1000 mg IV on days 1 and 15. Administer subsequent courses every 24 weeks or based on clinical evaluation but not sooner than every 16 weeks.
Methylprednisolone (100 mg IV, or its equivalent) 30 minutes prior to each infusion is recommended to reduce the incidence and severity of infusion reactions.
In a phase II trial, 161 patients with active rheumatoid arthritis (RA) despite methotrexate therapy were randomized to one of 4 groups:
methotrexate weekly (control);
rituximab;
rituximab plus cyclophosphamide (750 mg IV on days 3 and 17); or
rituximab plus methotrexate weekly.
All patients randomized to rituximab received 1000 mg IV on days 1 and 15.
In all groups treated with rituximab, a significantly higher proportion of patients had a 20% improvement in disease symptoms according to the ACR criteria at week 24 (65% to 76% vs. 38%; p <= 0.025).
The proportion of patients with 50% improvement in disease symptoms according to ACR criteria at 24 weeks, the primary end point, was significantly greater in the rituximab-methotrexate arm (43%) and the rituximab-cyclophosphamide arm (41%) as compared with methotrexate alone (13%).
These responses were maintained at week 48 in the rituximab-methotrexate group.
The majority of adverse events were associated with the first rituximab infusion.
Guidelines suggest adding or switching to an anti-TNF biologic, abatacept, or rituximab for patients with established disease and moderate or high disease activity after 3 months of methotrexate monotherapy or DMARD combination therapy.
Switching to a non-TNF biologic such as rituximab for patients with established disease who have a serious adverse event with a TNF blocker is recommended.
Further, a switch to a non-TNF biologic is an option for patients with moderate or high disease activity after >= 3 months of a TNF blocker or with a non-serious adverse event.
Also, a switch to another non-TNF biologic is an option for patients with moderate or high disease activity after >= 6 months of abatacept or with a non-serious adverse event to the drug.
Lastly, a switch to another type or category of non-TNF biologic is an option for patients with moderate or high disease activity after >= 6 months of the new drug. The goal is low disease activity or remission.
Pemphigus
rituximab + Corticosteroids
■ For the treatment of pemphigus vulgaris.
rituximab 1,000 mg IV every 2 weeks for 2 doses in combination with a tapering course of corticosteroids, initially.
Then, 500 mg IV at month 12 and every 6 months thereafter may be used based on clinical evaluation.
For relapsed disease, 1,000 mg IV once with consideration of resuming or increasing the corticosteroid dose based on clinical evaluation.
Consider Pneumocystis jirovecii pneumonia prophylaxis during and after rituximab treatment.
† For the treatment of pemphigus foliaceus.
Wegener's granulomatosis
■ For the treatment of Wegener's granulomatosis and microscopic polyangiitis in combination with glucocorticoids.
NOTE: Use of concomitant immunosuppressants other than corticosteroids has not been studied in patients with peripheral B-cell depletion after rituximab receipt.
† For the treatment of acute lymphocytic leukemia (ALL).
† For the treatment of relapsed or refractory acute lymphocytic leukemia (ALL) in combination with ifosfamide, etoposide, and carboplatin.
† For the treatment of refractory autoimmune hemolytic anemia.
† For steroid-refractory ITP.
† For previously untreated ITP in combination with dexamethasone.
† For the treatment of relapsing or refractory thrombotic thrombocytopenia purpura (TTP).
† For the treatment of acquired blood factor deficiency† in hemophilia A.
†
For the treatment of multicentric Castleman disease (MCD) associated with human herpesvirus 8 (HHV-8) infection† in HIV-infected patients.
† For the treatment of relapsing-remitting multiple sclerosis (RRMS).
† For the treatment of Burkitt's lymphoma in combination with Hyper-CVAD chemotherapy.
† For the treatment of steroid-refractory chronic graft-versus-host disease (GVHD).
† For the treatment of lupus nephritis† in patients with systemic lupus erythematosus (SLE) unresponsive to conventional therapy.
† For the treatment of newly diagnosed Waldenstrom macroglobulinemia, in combination with dexamethasone and cyclophosphamide.
† For the treatment of newly diagnosed Waldenstrom macroglobulinemia, in combination with bortezomib and dexamethasone.
Note: All patients received premedication with acetaminophen 1,000 mg PO and diphenhydramine 50 mg IV prior to rituximab and herpes zoster prophylaxis with valacyclovir or acyclovir.
† For the treatment of Waldenstrom macroglobulinemia, in combination with ibrutinib.
brentuximab vedotin
CD30-directed antibody-drug conjugate
Used as consolidation therapy in patients with high-risk classical Hodgkin lymphoma (cHL) and for the treatment of relapsed cHL, relapsed systemic anaplastic large-cell lymphoma (ALCL), and relapsed primary cutaneous pcALCL or CD30-expressing mycosis fungoides
JC virus infection resulting in progressive multifocal leukoencephalopathy and death has occurred
Classical Hodgkin lymphoma
NOTE: Brentuximab has been designated an orphan drug by the FDA for this indication.
■ For the treatment of classical Hodgkin lymphoma, after failure of autologous hematopoietic stem cell transplant (auto-HSCT) or after failure of at least 2 prior multi-agent chemotherapy regimens in patients who are not auto-HSCT candidates.
brentuximab vedotin 1.8 mg/kg IV (not to exceed 180 mg/dose) over 30 minutes every 3 weeks until disease progression.
Interrupt the infusion in patients who develop anaphylaxis or other infusion-related reactions.
Premedicate (e.g., acetaminophen, antihistamine, and/or corticosteroid) prior to subsequent infusions in patients who have previously experienced an infusion-related reaction.
Therapy interruption, dose reduction, and/or discontinuation may be necessary in patients who develop toxicity.
In a single-arm, multinational, phase II study (n = 102), treatment with up to 16 doses of brentuximab vedotin (median of 9 doses) led to an objective response rate assessed by independent central review (primary endpoint) of 75% in patients who had relapsed or refractory Hodgkin lymphoma after an autologous stem cell transplant (age range, 15 to 77 years; median of 3.5 prior systemic therapies).
The complete remission (CR) rate was 34% in this study.
The median time to objective response was 5.7 weeks (range, 5.1 to 56 weeks); the median response duration was 6.7 months. The median time to CR was 12 weeks (range, 5.1 to 56 weeks);
the median duration of CR was 20.5 months. Additionally, the median progression-free survival (PFS) time was 5.6 months; the estimated 12-month overall survival (OS) rate was 89%.
Eight patients in this study (6 patients in CR and 2 patients in partial remission) subsequently received an allogeneic stem cell transplant.
At a median follow-up time of 33.3 months (range, 1.8 to 57.3 months), the investigator-reported median response duration was 11.2 months, the estimated median PFS time was 9.3 months, and the estimated median OS time was 40.5 months.
■ For the treatment of classical Hodgkin lymphoma, as consolidation therapy following an autologous hematopoietic stem cell transplant in patients at high risk for relapse or progression.
brentuximab vedotin 1.8 mg/kg IV (not to exceed 180 mg/dose) over 30 minutes repeated every 3 weeks until disease progression or a maximum of 16 cycles;
start within 4 to 6 weeks after an autologous hematopoietic stem cell transplantation (auto-HSCT) or upon recovery after an auto-HSCT.
Interrupt the infusion in patients who develop anaphylaxis or other infusion-related reactions.
Premedicate (e.g., acetaminophen, antihistamine, and/or corticosteroid) prior to subsequent infusions in patients who have previously experienced an infusion-related reaction.
Therapy interruption, dose reduction, and/or discontinuation may be necessary in patients who develop toxicity.
brentuximab vedotin + Chemotherapy
■ For the treatment of previously untreated stage III or IV classical Hodgkin lymphoma, in combination with chemotherapy.
brentuximab vedotin 1.2 mg/kg (not to exceed 120 mg/dose) IV over 30 minutes repeated every 2 weeks until maximum of 12 doses, disease progression, or unacceptable toxicity in combination with doxorubicin 25 mg/m2 IV, vinblastine 6 mg/m2 IV, and dacarbazine 375 mg/m2 IV each given on days 1 and 15 repeated every 28 days for up to 6 cycles was evaluated in a randomized, phase III trial.[62863]
Administer primary prophylaxis with a granulocyte colony-stimulating factor starting in cycle 1 due to the high incidence of febrile neutropenia.
Interrupt the infusion in patients who develop anaphylaxis or other infusion-related reactions.
Premedicate (e.g., acetaminophen, antihistamine, and/or corticosteroid) prior to subsequent infusions in patients who have previously experienced an infusion-related reaction.
Therapy interruption, dose reduction, and/or discontinuation may be necessary in patients who develop toxicity.
BOXED WARNING
Progressive multifocal leukoencephalopathy
Fatal cases of progressive multifocal leukoencephalopathy (PML), caused by the John Cunningham virus (JC virus), have been reported with brentuximab therapy; some cases occurred within the first 3 months of starting therapy. Patients with prior immunosuppressive therapies or immunosuppressive disease may be at increased risk of JC virus infection and PML. Evaluate patients who develop new neurological, cognitive, or behavioral signs and symptoms such as changes in mood or behavior; confusion; memory impairment; changes in vision, speech, or walking; and/or decreased strength or weakness on one side of the body. Hold therapy if PML is suspected; discontinue brentuximab in patients with confirmed PML.
■ For the treatment of systemic anaplastic large-cell lymphoma after failure of at least 1 prior multi-agent chemotherapy regimen.
Note: Brentuximab vedotin has been designated an orphan drug by the FDA for the treatment of anaplastic large-cell lymphoma.
brentuximab vedotin 1.8 mg/kg (not to exceed 180 mg/dose) IV over 30 minutes repeated every 3 weeks until disease progression.
Interrupt the infusion in patients who develop anaphylaxis or other infusion-related reactions.
Premedicate (e.g., acetaminophen, antihistamine, and/or corticosteroid) prior to subsequent infusions in patients who have previously experienced an infusion-related reaction.
Therapy interruption, dose reduction, and/or discontinuation may be necessary in patients who develop toxicity.
In a single-arm, multicenter, phase II study (n = 58), treatment with up to 16 doses of brentuximab vedotin (median of 7 doses) led to an overall objective response rate (primary endpoint) of 86% in patients with relapsed or refractory systemic anaplastic large cell lymphoma (age range, 14 to 76 years; median of 2 prior systemic therapies).
The complete remission (CR) rate was 57% in this study. The median time to objective response was 5.9 weeks (range, 4.3 to 14 weeks); the median duration of objective response was 12.6 months.
The median time to CR was 11.9 weeks (range, 5.1 to 50.3 weeks); the median duration of CR was 13.2 months.
Additionally, the median progression-free survival time was 13.3 months and the estimated 12-month overall survival rate was 70%.
Of the 33 patients in this study who achieved a CR, 6 patients received a subsequent allogeneic stem cell transplant and 5 patients received a subsequent autologous stem cell transplant.
■ For the treatment of cutaneous T-cell lymphoma (CTCL), specifically mycosis fungoides or primary cutaneous anaplastic large-cell lymphoma, in patients who have received prior systemic therapy.
Note: Brentuximab vedotin has been designated an orphan drug by the FDA for the treatment of CTCL and mycosis fungoides.
brentuximab vedotin 1.8 mg/kg (not to exceed 180 mg/dose) IV over 30 minutes repeated every 3 weeks until disease progression or a maximum of 16 cycles.
Interrupt the infusion in patients who develop anaphylaxis or other infusion-related reactions.
Premedicate (e.g., acetaminophen, antihistamine, and/or corticosteroid) prior to subsequent infusions in patients who have previously experienced an infusion-related reaction.
Therapy interruption, dose reduction, and/or discontinuation may be necessary in patients who develop toxicity.
Cisplatin
Before administration:
Adequate hydration for 12 hours prior to cisplatin.
Prehydration with 2 L of an appropriate IV solution, and posthydration 2.5 L/m2/24 hrs.
0.9% Sodium Chloride 250 mL/hour for 2—4 hours prior to and post cisplatin.
OR post cisplatin 100-200 mL/hour for 6-12 hours.
(Patient should drink large quantities of liquids for 24 hours after Cisplatin infusion.)
Administration:
Cisplatin in 1L NS IV infusion over 3 hrs, OR
Cisplatin in 2L D5/Half-Saline IV infusion over 6 hrs
Maintenance of urine output of 100—150 mL/hour for 24 hours following cisplatin are recommended.
Forced diuresis if urine output < 100—150 mL/hour: 37.5g Mannitol as a 10% solution (37.5g Mannitol in 375 ml solution), or by administration of a diuretic if the kidney functions are normal.
Administration of Mannitol or a diuretic is also required if Cisplatin dose is higher than 60 mg/m2.
Premedication with antiemetics including a serotonin antagonist and corticosteroid is required to prevent severe nausea and vomiting.
Metoclopramide 2 mg/kg IV one-half hour before Cisplatin administration, and 1.5, 3.5, 5.5, and 8.5 hours after Cisplatin administration.
Aluminum needles, or IV sets containing aluminum should not be used for cisplatin preparation or administration because aluminum reacts with cisplatin to form a precipitate, causing loss of potency.
Following initial entry into the Platinol-AQ vial, the remaining solution is stable for 28 days protected from light or 7 days under fluorescent room light.
Visually inspect parenteral products for particulate matter and discoloration prior to administration whenever solution and container permit.
A minimum creatinine clearance of 50 mL/min is recommended prior to full dose cisplatin therapy.
Serum creatine < 1.5 mg/dl
Urea < 25 mg/dl
WBC > 4000/ul
Platelets > 100,000/ul
Audiogram: Within normal range.
Cisplatin may be diluted in 0.9% Sodium Chloride to a final concentration of 50—500 mg/L.
(These solutions are stable up to 24 hours at room temperature.)
Cisplatin is compatible in solutions containing an adequate amount of chloride ions and is incompatible with solutions having low concentrations of these ions.
Infuse over 6—8 hours. Generally, cisplatin should be given no faster than 1 mg/min.
MAXIMUM DOSAGE
The suggested maximum tolerated dose (MTD) for cisplatin is dependent on performance status, other chemotherapy agents or radiation given in combination, and disease state. The dosing of cisplatin may vary from protocol to protocol. If questions arise, clinicians should consult the appropriate references to verify the dose.
Adults
100 mg/m2 IV. Any dose above 100 mg/m2 should be confirmed with the prescriber and supported by appropriate documentation.
DOSING CONSIDERATIONS
Hepatic Impairment
No dosage adjustments are needed in patients with hepatic impairment.
Renal Impairment
No specific dosage guidelines have been established for cisplatin in patients with renal impairment. The manufacturer considers cisplatin contraindicated in patients with preexisting renal impairment, defined as a CrCl <= 50 mL/min. The risks versus the benefits of cisplatin must be considered prior to administering cisplatin to a patient with renal dysfunction. In a review of dosing guidelines, the following adjustments to cisplatin dosage have been recommended:
CrCl 46—60 mL/min: Decrease dose by 25%.
CrCl 31—45 mL/min: Decrease dose by 50%.
CrCl <= 30 mL/min: Not recommended; use an alternative drug.
Creatinine clearance calculation
Creatinine clearance = (140-age) x weight(kg)/(72 x serum cr(mg/dl)
(The resulting value is multiplied by a constant of 0.85 if the patient is female.)
ADMINISTRATION
For storage information, see the specific product information in the How Supplied section.
CAUTION: Observe and exercise appropriate precautions for handling, preparing, and administering cytotoxic drugs.
Injectable Administration
Cisplatin is administered intravenously but also may be administered intra-arterially.
Adequate renal and hematologic function should be established prior to administering cisplatin.
To decrease the incidence and severity of nephrotoxicity, hydration consisting of 0.9% Sodium Chloride 250 mL/hour for 2—4 hours prior to and post cisplatin, and maintenance of urine output of 100—150 mL/hour for 24 hours following cisplatin are recommended.
A minimum creatinine clearance of 50 mL/min is recommended prior to full dose cisplatin therapy.
Premedication with antiemetics including a serotonin antagonist and corticosteroid is required to prevent severe nausea and vomiting.
Aluminum needles, or IV sets containing aluminum should not be used for cisplatin preparation or administration because aluminum reacts with cisplatin to form a precipitate, causing loss of potency.
Following initial entry into the Platinol-AQ vial, the remaining solution is stable for 28 days protected from light or 7 days under fluorescent room light.
Visually inspect parenteral products for particulate matter and discoloration prior to administration whenever solution and container permit.
Intravenous Administration
Reconstitution:
NOTE: Cisplatin lyophilized powder (Platinol) is no longer available; however, the manufacturer has a procedure to make the powder available for patients for whom Platinol-AQ is not a medically acceptable substitute. Requests can be made by calling 1—800—437—0994.
Platinol powder for injection 10 or 50 mg is reconstituted with 10 or 50 mL, respectively, of sterile water for injection to give solutions containing 1 mg/mL of cisplatin.
Following reconstitution with sterile water for injection, cisplatin 1 mg/mL is stable for 20 hours at 20 degrees C. When reconstituted with bacteriostatic water, cisplatin 1 mg/mL solutions are stable for 72 hours under refrigeration. A precipitate will form upon refrigeration but will go back into solution without loss of potency upon warming.
Non-small cell lung cancer (NSCLC)
GoTo Lung Ca Regimens below.
Head and neck cancer
GoTo Systemic Treatment for Head & Neck Cancers
Bladder Cancer
Cisplatin monotherapy
■ For the treatment of bladder cancer as a single agent.
50 to 70 mg/m2 IV as a single dose once every 3 to 4 weeks depending on the extent of prior exposure to radiation therapy and/or prior chemotherapy.
50 mg/m2 IV once every 4 weeks is recommended in heavily pretreated patients.
MVAC regimen
† Neoadjuvant treatment of muscle-invasive bladder cancer.
(
Vinblastine (3 mg/m2 IV on days 2, 15, and 22),
Methotrexate (30 mg/m2 slow IV push on days 1, 15, and 22),
Adriamycin/doxorubicin (30 mg/m2 slow IV push on day 2).
MVAC regimen
† First-line treatment of advanced or metastatic bladder cancer
(MVAC regimen) every 28 days for up to 6 cycles
Cisplatin (70 mg/m2 IV on day 2),
Vinblastine (3 mg/m2 IV on days 2, 15, and 22),
Methotrexate (30 mg/m2 slow IV push on days 1, 15, and 22),
Adriamycin/doxorubicin (30 mg/m2 slow IV push on day 2).
*Has been evaluated in patients with advanced or metastatic transitional cell carcinoma of the bladder in a long-term analysis of a multicenter, randomized, phase III trial.
MVAC regimen + G-CSF
(MVAC regimen) every 28 days
Cisplatin 70 mg/m2 IV on day 1
vinblastine (3 mg/m2 IV on days 1, 15, and 22)
methotrexate (30 mg/m2 IV on days 1, 15, and 22)
Adriamycin/doxorubicin (30 mg/m2 IV on day 1).
All patients in this study received granulocyte colony-stimulating factor (G-CSF) following chemotherapy.
*Was studied in another randomized, phase III trial.
Cisplatin + Gemcitabine (GC regimen) vs MVAC regimen
† First-line treatment of locally advanced or metastatic transitional-cell bladder cancer
1. Cisplatin + Gemcitabine (GC regimen) repeated every 28 days for up to 6 cycles.
Cisplatin 70 mg/m2 on day 2
Gemcitabine 1,000 mg/m2 IV over 30 to 60 minutes on days 1, 8, and 15
VS
2. MVAC regimen
In a multicenter, randomized, phase III trial. In a long-term analysis of this study, GC led to nonsignificantly different overall survival (OS) (14 vs. 15.2 months; adjusted hazard ratio (HR) = 0.99; 95% CI, 0.79 to 1.23) and progression-free survival (PFS) (7.7 vs. 8.3 months; adjusted HR = 1.01; 95% CI, 0.81 to 1.25) times compared with MVAC. The 5-year OS (13% vs. 15.3%) and PFS (9.8% vs. 11.3%) rates were nonsignificantly lower with GC compared with MVAC; however, GC was associated with significantly less grade 3 or 4 neutropenic sepsis (1% vs. 12%; p < 0.001) and mucositis (1% vs. 22%; p = 0.001). Treatment-related death was also reported less often in the GC arm (1% vs. 3%).
Testicular cancer
■ For the treatment of testicular cancer.
BEP regimen regimen and PVB regimen
Adults and Children
Cisplatin 20 mg/m2 IV as a single dose once daily for 5 days with bleomycin and etoposide (BEP regimen); repeat every 3 weeks for 2 cycles or more. Cisplatin is also given in combination with vinblastine and bleomycin (PVB regimen). Patients receiving the etoposide containing regimen have less neuropathies than those receiving the regimen containing vinblastine.
Penile cancer
† For the treatment of locally advanced or metastatic penile cancer
Cisplatin + methotrexate + bleomycin
Cisplatin 75 mg /m2 IV on day 1 in combination with methotrexate 25 mg/m2 IV bolus on days 1 and 8 and bleomycin 10 units/m2 IV on days 1 and 8, repeated every 21 days.
Treatment was given for 6 cycles if a complete remission was achieved. Patients who achieved stable disease or a partial response, continued treatment until disease progression. Bleomycin was discontinued after a maximum cumulative dose of 200 units/m2 was given.
† For the neoadjuvant treatment of locally advanced or metastatic carcinoma of the penis.
Cisplatin + paclitaxel + ifosfamide
Cisplatin 25 mg/m2/day IV over 2 hours on days 1, 2, 3 in combination with paclitaxel 175 mg/m2 IV over 3 hours on day 1 and ifosfamide 1200 mg/m2/day IV over 2 hours on days 1, 2, 3, repeated every 3 to 4 weeks.
Mesothelioma
■ 1. For the treatment of unresectable malignant pleural mesothelioma
Cisplatin + Pemetrexed
Cisplatin + Pemetrexed. Repeat every 21 days.
Pemetrexed 500 mg/m2 IV over 10 minutes.
Cisplatin 75 mg/m2 IV over 2 hours on day 1, beginning approximately 30 minutes after the end of the pemetrexed infusion.
Premedicate pemetrexed with Dexamethasone 4 mg by mouth twice daily for 3 days, beginning the day before pemetrexed administration to reduce cutaneous reactions.
Folic acid (400 to 1,000 mcg by mouth daily) and vitamin B12 (1 mg IM every 3 months) beginning 7 days prior to the first dose of pemetrexed and continuing for 21 days after the last dose to reduce the severity and frequency of hematologic and GI toxicities.
(After the first dose, vitamin B12 may be given on the same day as pemetrexed. Do not substitute oral for IM vitamin B12.)
Pemetrexed plus cisplatin for chemotherapy-naive patients with malignant mesothelioma (n = 226) resulted in a median survival time of 12.1 months compared with 9.3 months for recipients of cisplatin monotherapy (n = 222) (HR 0.77; p = 0.02); approximately 75% of patients had either stage 3 or 4 disease. Of patients who received supplementation with folic acid and vitamin B12, the median overall survival was 13.2 months vs. 9.4 months, respectively (HR 0.71; p = 0.022). The median time to progressive disease in the intent-to-treat group (regardless of supplementation) was 5.7 months in the pemetrexed group and 3.9 months in those who received cisplatin monotherapy (HR 0.68; p = 0.001), and the tumor response rate was 41.3% compared with 16.7%, respectively (p < 0.001).
† 2. For the treatment of malignant mesothelioma.
Cisplatin + Gemcitabine
(GC regimen) every 28 days for 6 cycles.
Cisplatin 100 mg/m2 IV on day 1
Gemcitabine 1000 mg/m2 IV on days 1, 8 and 15. ((IV over 30 to 60 minutes)).
Alternately, cisplatin 80 mg/m2 IV on day 1 in combination with gemcitabine 1250 mg/m2 IV on days 1 and 8, every 21 days for 6 cycles, has also been studied.
† 3. For the treatment of malignant peritoneal mesothelioma
Intraperitoneal Cisplatin and mitomycin C
Four to 6 L of isotonic dialysis fluid was circulated at a flow rate of 500 to 700 mL/min and heated to achieve an intraperitoneal temperature between 42 to 45 degrees C.
Intraperitoneal chemotherapy with mitomycin 0.5 mg/kg in combination with cisplatin 0.7 mg/kg was administered over 90 minutes.
5-year overall survival was 28.9%; median overall survival was 35.6 months.
Ovarian cancer
■ For the treatment of ovarian cancer.
Cisplatin + cyclophosphamidee
Cisplatin 50 to 60 mg/m2 IV as a single dose once every 21 days in combination with cyclophosphamide.
■ For the treatment of advanced ovarian cancer.
Cisplatin + paclitaxel
Cisplatin 75 mg/m2 IV as a single dose once every 21 days in combination with paclitaxel.
This combination is associated with an increased survival as compared to cisplatin + cyclophosphamide.
† First-line treatment of optimally debulked, stage III ovarian cancer.
Intravenous paclitaxel + Intraperitoneal Cisplatin + intraperitoneal paclitaxel
Paclitaxel 135 mg/m2 intravenously over 24 hours on day 1
cisplatin 100 mg/m2 intraperitoneally on day 2
paclitaxel 60 mg/m2 intraperitoneally on day 8.
Administer every 3 weeks for 6 cycles.
Extended overall survival and progression free survival in a phase III trial compared to cisplatin IV/paclitaxel IV.
Endometrial cancer
† For the treatment of advanced or recurrent endometrial cance.
Cisplatin + doxorubicin + paclitaxel (TAP regimen) + Filgrastim
Cisplatin 50 mg/m2 IV on day 1, given immediately after doxorubicin (45 mg/m2) on day 1, then administer paclitaxel (160 mg/m2) on day 2; give every 21 days.
Filgrastim (5 mcg/kg) was administered on days 3—12.
In clinical trials treatment was continued for up to 7 cycles or until disease progression.
A phase III trial showed an increase in response rate, progression-free survival, and overall survival in patients receiving paclitaxel, doxorubicin, and cisplatin (TAP) vs. cisplatin and doxorubicin alone. Thrombocytopenia and neuropathy were higher in the TAP arm.
Carboplatin (Paraplatin)
Carboplatin (Paraplatin)
Platinum alkylating antineoplastic agent; on a molar basis, carboplatin is 45 times less cytotoxic than cisplatin; as effective as cisplatin in ovarian, non-small cell and small cell lung cancers; not recommended for routine treatment of testicular or head and neck cancers.
Vomiting occurs frequently with carboplatin therapy; ensure that patients are premedicated with appropriate antiemetic therapy prior to initiating therapy with carboplatin.
SUPPLIED: Carboplatin/Paraplatin Intravenous Inj Sol: 1mL, 10mg
MAXIMUM DOSAGE
The suggested maximum tolerated dose (MTD) for carboplatin is dependent on performance status, other chemotherapy agents or radiation given in combination, and disease state. The dosing of carboplatin may vary from protocol to protocol. If questions arise, clinicians should consult the appropriate references to verify the dose.
Adults, Geriatric
Maximum dosage information is not available. For doses calculated using the Calvert formula with an estimated GFR, it is recommended to use a maximum GFR = 125 mL/min in the calculation.
Adolescents, Children, Infants, Neonates
Safety and efficacy have not been established. Maximum dosage information is not available. For doses calculated using the Calvert formula with an estimated GFR, it is recommended to use a maximum GFR = 125 mL/min in the calculation.
DOSING CONSIDERATIONS
Hepatic Impairment
No dosage adjustments are needed in patients with hepatic impairment.
Renal Impairment
NOTE: Recommendations are only available for the initial course of treatment for ovarian cancer with carboplatin as a single agent or with cyclophosphamide. Subsequent adjustments should be done base on the toxicity of the previous course.
CrCl >= 60 ml/min: no dosage adjustment needed.
CrCl 41—59 ml/min: give 250 mg/m2 on day 1 of the initial course for ovarian cancer.
CrCl 16—40 ml/min: give 200 mg/m2 on day 1 of the initial course for ovarian cancer.
CrCl <= 15 ml/min: Data too limited to permit a recommendation.
Creatinine clearance calculation
Creatinine clearance = (140-age) x weight(kg)/(72 x serum cr(mg/dl)
(The resulting value is multiplied by a constant of 0.85 if the patient is female.)
Creatinine clearance = Estimated GFR
Carboplatin AUC Dose Calculation (Calvert formula)
Carboplatin Dose = Target AUC x (GFR + 25)
GoTo Medscape ONLINE Carboplatin AUC Dose Calculation (Calvert formula)
GoTo MedicineWorld.Org ONLINE Carboplatin dose calculator
Above two calculators use the following formula for calculation of carboplatin dose.
First of all the creatinine clearance is calculated using the modified modified Cockcroft-Gault formula which is given below:
Creatinine clearance = (140 - age) x actual weight(kg) / (72 x serum creatinine(mg/dl))x gender correction factor (1 for male 0.85 for female)
Carboplatin dose is calculated using the following formula:
Carboplatin dose = target AUC(mg/ml/min) x (CrCl + 25)(ml/min)
GoTo GlobalRPh ONLINE for alternate calculation Carboplatin AUC Calculator for Carboplatin Dosing.
Relevant package insert data:
Previously treated patients: a target AUC of 4-6 mg/mL•min using single agent Carboplatin Inj appears to provide the most appropriate dose range. For patients who previously DID NOT receive chemotherapy (untreated), a target AUC of 7 (range: 6-8) mg/mL per minute has been recommended when carboplatin is used alone.
Dose Adjustment Recommendations: Pretreatment platelet count and performance status are important prognostic factors for severity of myelosuppression in previously treated patients. The suggested dose adjustments for single agent or combination therapy shown in the table below are modified from controlled trials in previously treated and untreated patients with ovarian carcinoma. Blood counts were done weekly, and the recommendations are based on the lowest post-treatment platelet or neutrophil value.
| Platelets | Neutrophils | Adjusted Dose* (From Prior Course) |
| >100,000 | >2000 | 125% |
| >50-100,000 | >500-2000 | No Adjustment |
| <50,000 | <500 | 75% |
Intermittent hemodialysis
Multiple reports document the use of carboplatin in patients who are receiving concurrent hemodialysis. Most of the reports indicate that carboplatin is removed by hemodialysis. Studies indicate that anuric patients may receive carboplatin at initial doses not exceeding 150 mg/m2 IV followed by dialysis within 24—48 hours of treatment (written communication, Bristol-Myers Squibb Oncology/Immunology Division, September 1999).
ADMINISTRATION
CAUTION: Observe and exercise appropriate precautions for handling, preparing, and administering cytotoxic drugs.
Aluminum needles or IV sets containing aluminum should not be used for carboplatin preparation or administration because aluminum reacts with carboplatin to form a precipitate, causing loss of potency.
Injectable Administration
Carboplatin is administered intravenously as an infusion.
Routine hydration is not required with carboplatin therapy. Hydration should be considered in patients with renal impairment or in those receiving concurrent nephrotoxic agents.
Visually inspect parenteral products for particulate matter and discoloration prior to administration whenever solution and container permit.
Intravenous Administration
Reconstitution of vials:
Reconstitute carboplatin 50, 150, or 450 mg vials with 5, 15, or 45 mL, respectively, of sterile water for injection, 5% Dextrose for injection, or sodium chloride injection. The reconstituted vials should have a concentration of 10 mg/mL of carboplatin.
Reconstituted vials are stable for 24 hours at room temperature (25 degrees C). Paraplatin multidose (10 mg/mL) vials are stable for up to 14 days following initial entry into the vial.
Further dilution for infusion:
Further dilute carboplatin solution (10 mg/mL) with 5% Dextrose injection or 0.9% Sodium Chloride injection to concentrations of 0.5—4 mg/mL.
Carboplatin solutions further diluted with 5% Dextrose injection or 0.9% Sodium Chloride injection are stable for 8 hours at room temperature (25 degrees C). Since carboplatin solutions are preservative-free the manufacturer recommends discarding any unused carboplatin solution after 8 hours.
Intermittent infusion:
Infuse appropriate dose IV over 15 minutes to 1 hour.
Continuous infusion:
Infuse at a rate to allow administration of the entire dose over a 24 hour period.
Other Administration Route(s)
Intraperitoneal Administration †:
NOTE: Carboplatin is not approved by the FDA for intraperitoneal administration.
Add carboplatin to a pre-warmed sterile 0.9% Sodium Chloride solution prior to instilling (i.e., body-temperature).
Dilute carboplatin dosage in 2 liters 0.9% Sodium Chloride injection, instill and dwell for 2—4 hours, then drain by gravity as completely as possible.
Carboplatin is administered via a Tenckhoff catheter or a percutaneously inserted peritoneal dialysis catheter.
STORAGE
Generic:
- Protect from light
- Store at 77 degrees F; excursions permitted to 59-86 degrees F
Paraplatin:
- Discard product if it contains particulate matter, is cloudy, or discolored
- Product is stable for up to 14 days at 77 degrees F following multiple uses
- Protect from light
- Store at controlled room temperature (between 68 and 77 degrees F)
- Store in carton until time of use
† For the treatment of advanced squamous cell carcinoma of the head and neck in combination with paclitaxel and radiotherapy.
carboplatin/paclitaxel/Radiation
carboplatin 100 mg/m2 IV weekly in combination with paclitaxel (40 to 45 mg/m2 IV weekly).
Chemotherapy was administered weekly prior to radiation therapy.
In a clinical trial, 62 patients were administered carboplatin-paclitaxel concomitantly with radiation therapy.
An overall survival of 33 months was achieved.
A complete response (CR) occurred in 75% of patients; among patients with a CR, an overall survival of 49 months was achieved. At a follow-up of 30 months, a local control rate of 63% was observed.
Preoperative chemoradiotherapy for esophageal or junctional cancer.
Chemoradiotherapy regimen
carboplatin (AUC of 2 mg per milliliter per minute) and paclitaxel (50 mg/M2)
Weekly administration for 5 weeks and concurrent radiotherapy (41.4 Gy in 23 fractions, 5 days per week),
† For the first-line treatment of ovarian cancer in combination with paclitaxel.
carboplatin/paclitaxel/Radiation
carboplatin AUC 5 to 7.5 IV on day 1 in combination with paclitaxel (175 to 185 mg/m2 IV over 3 hours on day 1), every 3 weeks for 6 cycles.
In clinical trials, carboplatin combined with paclitaxel has been shown to be less toxic and produce similar efficacy to cisplatin combined with paclitaxel as first-line treatment of patients with advanced ovarian cancer.
Additionally, carboplatin AUC 6 IV on day 1 in combination with paclitaxel (80 mg/m2 IV on days 1, 8, and 15), every 3 weeks for 6 cycles has been given.
This dose-dense combination was compared to conventional carboplatin/paclitaxel in 631 patients with advanced ovarian cancer. Progression-free survival, the primary endpoint, was significantly higher in the dose-dense arm (28 months vs. 17.2 months, p = 0.0015).
Non-small cell lung cancer (NSCLC) treatment regimens †
Carboplatin + Paclitaxel
† For the treatment of advanced or metastatic NSCLC.
Carboplatin AUC 6 IV on day 1 in combination with paclitaxel 200 mg/m2 IV on day 1 given every 21 days.
Produced an overall survival of 12.3 months in a phase III comparison of 4 chemotherapy doublets in advanced NSCLC.
In another similar 4 arm phase III comparison, carboplatin AUC 6 IV on day 1 in combination with paclitaxel 225 mg/m2 IV on day 1 given every 21 days, produced an overall survival of 7.8 months, which was similar to the reference regimen of cisplatin and paclitaxel.
Carboplatin + gemcitabine
† For first-line treatment of inoperable, locally advanced or metastatic non-small cell lung cancer (NSCLC).
AUC 5 IV on day 1 in combination with gemcitabine 1,200 mg/m2 IV on days 1 and 8, every 21 days for 6 cycles.
Alternatively, carboplatin AUC 5 IV on day 1 with gemcitabine 1,000 mg/m2 IV on days 1, 8 and 15, repeated every 28 days for 4 cycles or,
carboplatin AUC 5 IV day 1 with gemcitabine 1,000 mg/m2 IV on days 1 and 8, repeated every 21 days for 4 cycles, have also been studied.
Carboplatin + docetaxel
† For first-line treatment of unresectable, locally advanced or metastatic non-small cell lung cancer (NSCLC).
AUC 6 IV on day 1 in combination with docetaxel (75 mg/m2 IV) on day 1 given every 21 days for 4 to 6 cycles.
In a phase III trial of 1,203 patients with unresectable locally advanced or metastatic NSCLC, docetaxel and platinum combinations (cisplatin or carboplatin) were compared with cisplatin/vinorelbine.
No difference was observed between docetaxel/carboplatin and cisplatin/vinorelbine in overall survival, the primary endpoint.
Grade 3 and 4 anemia and nausea/vomiting were significantly lower in both docetaxel containing arms.
In addition, hospitalizations and treatment discontinuation secondary to toxicity were higher with cisplatin/vinorelbine.
A separate phase III trial conducted in 422 patients with inoperable, locally advanced or metastatic NSCLC compared docetaxel/carboplatin to mitomycin C/cisplatin plus either ifosfamide or vinblastine.
The primary endpoint, 1-year overall survival, was not significantly different between the treatment arms.
Grade 3 and 4 neutropenia, infection, and mucositis were all significantly higher with docetaxel/carboplatin, while quality of life scores were significantly better.
Carboplatin + pemetrexed
† For the first-line treatment of stage IIIB or IV non-small cell lung cancer (NSCLC).
AUC 5 IV on day 1 in combination with pemetrexed (500 mg/m2 IV on day 1), repeated every 3 weeks for 4 cycles.
In a phase III trial, 436 patients were randomized to pemetrexed/carboplatin or gemcitabine/carboplatin.
No significant differences were observed for the primary endpoint, health-related quality of life, or the secondary endpoint, overall survival.
Grade 3 or 4 toxicities were significantly worse in the gemcitabine/carboplatin arm.
Pemetrexed + carboplatin
† For the first line treatment of stage IIIB or IV non-small cell lung cancer (NSCLC).
Pemetrexed 500 mg/m2 IV on day 1 in combination with carboplatin (AUC 5 IV) on day 1, repeated every 3 weeks for up to 4 cycles;
after the initial dose, monitor the nadir ANC and platelet count to assess for possible pemetrexed dose reductions.
To prevent or minimize toxicities, all patients should also receive the following concomitant medications:
dexamethasone 4 mg PO twice daily for 3 consecutive days, beginning the day before each pemetrexed dose;
folic acid 400 to 1,000 mcg PO daily beginning 7 days prior to the first pemetrexed dose and continuing until 21 days after the last pemetrexed dose; and
vitamin B12 injection 1 mg IM one week prior to the first dose of pemetrexed and every 3 cycles (every 9 weeks) thereafter (do not substitute oral vitamin B12 for the IM injection). After the first vitamin B12 injection, subsequent injections may be given on the same day as pemetrexed treatment.
Pemetrexed/carboplatin vs gemcitabine/carboplatin
In a multicenter, randomized phase 3 trial, no significant differences were observed for the primary end point, health-related quality of life, or the secondary end point, overall survival for patients treated with pemetrexed/carboplatin compared with gemcitabine/carboplatin (n = 436);
grade 3 or 4 toxicities were significantly worse in the gemcitabine/carboplatin arm.
Carboplatin + nanoparticle albumin-bound paclitaxel
† For the first-line treatment of locally advanced or metastatic non-small cell lung cancer (NSCLC) in patients who are not candidates for curative surgery or radiation.
(NOTE: Nanoparticle albumin-bound paclitaxel is FDA approved in combination with carboplatin for the first-line treatment of locally advanced or metastatic NSCLC.)
AUC 6 IV on day 1 in combination with nanoparticle albumin-bound (nab) paclitaxel 100 mg/m2 IV over 30 minutes on days 1, 8, and 15 of each 21-day cycle;
administer carboplatin immediately after the nab-paclitaxel infusion on day 1.
The primary endpoint of overall response rate (ORR) assessed by independent radiologic review was significantly improved with nab-paclitaxel plus carboplatin (median number of cycles, 6; range, 1 to 31 cycles) compared with solvent-based (sb) paclitaxel plus carboplatin (33% vs. 25%; response rate ratio = 1.313; 95% CI, 1.082 to 1.593; p = 0.005) in patients with previously untreated non-resectable stage IIIB or stage IV non-small cell lung cancer (NSCLC) in a multicenter, randomized, phase III trial (n = 1,052).
All responding patients in the nab-paclitaxel arm had a partial response (PR); 1 patient in the sb-paclitaxel arm achieved a complete response, all others had a PR.
In a subgroup analysis, the ORR was significantly improved with nab-paclitaxel plus carboplatin in patients with squamous cell histology (41% vs. 24%; p < 0.001) but not nonsquamous cell histology (26% vs. 25%).
Although treatment with nab-paclitaxel plus carboplatin was not statistically superior compared with sb-paclitaxel plus carboplatin for the secondary endpoints of median progression-free survival (6.3 months vs. 5.8 months, HR = 0.902; 95% CI, 0.767 to 1.06) and overall survival (12.1 months vs. 11.2 months, HR = 0.922; 95% CI, 0.797 to 1.066) times, noninferiority was demonstrated.
Pemetrexed (Alimta)
Pemetrexed disodium (Alimta)
• Folic Acid Analogs or folate antimetabolites
• Pemetrexed is a folate analog inhibitor (antifolate) that disrupts folate-dependent metabolic processes essential for cell replication. In vitro, it inhibits folate-dependent enzymes involved in the de novo biosynthesis of thymidine and purine nucleotides, including thymidylate synthase (TS), dihydrofolate reductase (DHFR), and glycinamide ribonucleotide formyltransferase (GARFT). Inhibition of TS leads to a reduction in thymidine, which is needed for DNA synthesis. Additionally, inhibition of GARFT prevents de novo purine biosynthesis, leading to further depletion of required cell precursors. Thus, both thymidine and hypoxanthine (a purine) are needed to rescue a cell from cytotoxicity from pemetrexed.
• Like 5-fluorouracil and raltitrexed, pemetrexed primarily inhibits thymidylate synthase (TS) resulting in decreased thymidine available for DNA synthesis. Pemetrexed also inhibits dihydrofolate reductase (DHFR) and glycinamide ribonucleotide formyltransferase (GARFT), which are key enzymes required for the de novo bio-synthesis of
thymidine and purine nucleotides.
Once pemetrexed gains entry to the cell, through the reduced folate carrier, it is polyglutamated. Glutamation increases cellular retention and the intracellular half-life of pemetrexed, as well as making the polyglutamated metabolites greater than 60-fold more potent in their inhibition of TS.
Pemetrexed is a radiation-sensitizing agent. Pemetrexed induces cell cycle arrest in the G1/S phase.
• Used for the treatment of NSCLC and mesothelioma
Requires supplementation with folic acid and vitamin B12 to mitigate hematologic and GI toxicities, and dexamethasone for cutaneous toxicities
SUPPLIED As Alimta Intravenous Inj Pwd F/Sol: 100mg, 500mg
Reconstitution:
100 mg vial: Reconstitute with 4.2 mL of preservative-free 0.9% Sodium Chloride Injection, to achieve a concentration of 25 mg/mL.
500 mg vial: Reconstitute with 20 mL of preservative-free 0.9% Sodium Chloride Injection, to achieve a concentration of 25 mg/mL.
Do not use calcium-containing solutions for reconstitution.
Visually inspect parenteral products for particulate matter and discoloration prior to administration whenever solution and container permit. The reconstituted solution should be clear and colorless or a light yellow to yellow-green.
Further dilution is required; discard any unused contents left in the vial.
Store unreconstituted product at 77 degrees F; excursions permitted to 59-86 degrees F
Storage following reconstitution: Reconstituted vials may be stored in the refrigerator for up to 24 hours at 2 to 8 degrees C (36 to 46 degrees F).
Dilution:
Visually inspect parenteral products for particulate matter and discoloration; discard the vial if particulate matter is observed.
Withdraw the appropriate amount of pemetrexed from the reconstituted vial and dilute in preservative-free 0.9% Sodium Chloride Injection to a total volume of 100 mL.
Storage following dilution: The diluted admixture may be stored in the refrigerator for up to 24 hours (after reconstitution) at 2 to 8 degrees C (36 to 46 degrees F).
INDICATIONS AND USAGE:
• Locally Advanced or Metastatic Nonsquamous Non-Small Cell Lung Cancer:
• Initial treatment in combination with cisplatin.
• Maintenance treatment of patients whose disease has not progressed after four cycles of platinum-based first-line chemotherapy.
• After prior chemotherapy as a single agent.
• Mesothelioma: in combination with cisplatin.
Limitations of Use:
• ALIMTA is not indicated for the treatment of patients with squamous cell non-small cell lung cancer.
Laboratory Monitoring and Dose Reduction/Discontinuation Recommendations Monitoring
Complete blood cell counts, including platelet counts, should be performed on all patients receiving ALIMTA.
Patients should be monitored for nadir and recovery, which were tested in the clinical study before each dose and on days 8 and 15 of each cycle.
Patients should not begin a new cycle of treatment unless the ANC is ≥1500 cells/mm3, the platelet count is ≥100,000 cells/mm3, and creatinine clearance is ≥45 mL/min. Periodic chemistry tests should be performed to evaluate renal and hepatic function
Non-squamous non-small cell lung cancer treatment regimens
Pemetrexed monotherapy
For the treatment of recurrent, metastatic, non-squamous NSCLC, after prior chemotherapy.
(NOTE: Pemetrexed is not indicated for the treatment of patients with squamous cell NSCLC.)
Standard Pemetrexed regimen
Pemetrexed 500 mg/m2 IV over 10 minutes on day 1, every 21 days until disease progression or unacceptable toxicity;
after the initial dose, monitor the nadir ANC and platelet count to assess for possible pemetrexed dose reductions.
To prevent or minimize toxicities, all patients should also receive the following concomitant medications:
dexamethasone 4 mg PO twice daily for 3 consecutive days, beginning the day before each pemetrexed dose;
folic acid 0.4 mg (0.4mg-1mg) PO daily beginning 7 days prior to the first pemetrexed dose and continuing until 21 days after the last pemetrexed dose; and
vitamin B12 injection 1 mg IM one week prior to the first dose of pemetrexed and every 3 cycles (every 9 weeks) thereafter (do not substitute oral vitamin B12 for the IM injection).
After the first vitamin B12 injection, subsequent injections may be given on the same day as pemetrexed treatment.
Pemetrexed vs docetaxel
Overall survival (8.3 months vs. 7.9 months), progression-free survival (2.9 months vs. 2.9 months), and overall response rate (8.5% vs. 8.3%) were not significantly different in patients receiving treatment with pemetrexed compared with docetaxel
(in a multicenter, open-label, non-inferiority study of patients with recurrent stage III/IV NSCLC after prior treatment with one prior chemotherapy regimen for advanced disease.)
P2=P2=328: This a retrospective analysis of the large phase III study of previous treated patients with NSCLC Standard Pemetrexed vs docetaxel (75mg/m2
Patients with non-squamous histology treated with Pemetrexed had significantly better survival compared to all others on trial.
Doxetaxel had better survival than pemetrexed in the squamous subgroup.
The hypothesis for this observation is that thymidylate symthase.(TS) overexpression in squamous cell carcinoma leads to reduced sensitivity to pemetrexed.
Pemetrexed monotherapy
For maintenance treatment of patients with locally advanced or metastatic, non-squamous NSCLC, whose disease has not progressed after 4 cycles of first-line platinum-based chemotherapy.
Standard Pemetrexed regimen until disease progression or unacceptable toxicity.
Median progression-free survival (PFS) (4 months vs. 2 months) and overall survival (OS) (13.4 months vs. 10.6 months) were significantly improved following maintenance therapy with pemetrexed (median number of cycles, 5) compared with placebo
(in patients with stage IIIB or IV NSCLC who had not progressed after 4 cycles of platinum-based first line chemotherapy in a multicenter, double-blind, phase 3 clinical trial (n = 663).)
In a subgroup analysis, while OS (15.5 months vs. 10.3 months) and PFS (4.4 months vs. 1.8 months) were significantly improved in patients with non-squamous NSCLC treated with pemetrexed, there was not a significant effect on patients with squamous-cell NSCLC.
These results were consistent with the findings of another multicenter, double-blind, phase 3 trial (n = 539), where median PFS (4.1 months vs. 2.8 months) and OS (4.1 months vs. 2.8 months) were significantly improved following maintenance therapy with pemetrexed compared with placebo in patients with nonquamous, stage IIIB or IV NSCLC that had not progressed after 4 cycles of pemetrexed/cisplatin first-line chemotherapy.
Pemetrexed + cisplatin
For first-line treatment of locally advanced or metastatic, non-squamous, NSCLC.
Pemetrexed 500 mg/m2 IV over 10 minutes,
cisplatin (75 mg/m2 IV over 2 hours beginning approximately 30 minutes after the end of the pemetrexed infusion) on day 1, every 21 days for up to 6 cycles or until disease progression or unacceptable toxicity;
after the initial dose, monitor the nadir ANC and platelet count to assess for possible pemetrexed dose reductions.
To prevent or minimize toxicities, all patients should also receive the following concomitant medications: dexamethasone 4 mg PO twice daily for 3 consecutive days, beginning the day before each pemetrexed dose; folic acid 400 to 1,000 mcg PO daily beginning 7 days prior to the first pemetrexed dose and continuing until 21 days after the last pemetrexed dose; and
vitamin B12 injection 1 mg IM one week prior to the first dose of pemetrexed and every 3 cycles (every 9 weeks) thereafter (do not substitute oral vitamin B12 for the IM injection). After the first vitamin B12 injection, subsequent injections may be given on the same day as pemetrexed treatment.
Pemetrexed/cisplatin vs gemcitabine/cisplatin
Overall survival (10.3 months vs. 10.3 months), progression-free survival (4.8 months vs. 5.1 months), and overall response rate (27.1% vs. 24.7%) were not significantly different
(in chemotherapy naive stage IIIb/IV NSCLC patients randomized to treatment with pemetrexed plus cisplatin compared with gemcitabine plus cisplatin in a multicenter, open-label, non-inferiority study (n = 1,725);)
patients in both arms received folic acid, vitamin B12, and dexamethasone.
Analysis of NSCLC histology revealed significantly worse median survival for pemetrexed/cisplatin patients with squamous-cell histology compared with gemcitabine/cisplatin (9.4 months vs. 10.8 .
Grade 3 or 4 neutropenia (27% vs. 15%), anemia (10% vs. 6%), and thrombocytopenia (13% vs. 4%) were all significantly higher in the cisplatin/gemcitabine arm.
Pemetrexed + carboplatin
† For the first line treatment of stage IIIB or IV non-small cell lung cancer (NSCLC).
Pemetrexed 500 mg/m2 IV on day 1 in combination with carboplatin (AUC 5 IV) on day 1, repeated every 3 weeks for up to 4 cycles;
after the initial dose, monitor the nadir ANC and platelet count to assess for possible pemetrexed dose reductions.
To prevent or minimize toxicities, all patients should also receive the following concomitant medications:
dexamethasone 4 mg PO twice daily for 3 consecutive days, beginning the day before each pemetrexed dose;
folic acid 400 to 1,000 mcg PO daily beginning 7 days prior to the first pemetrexed dose and continuing until 21 days after the last pemetrexed dose; and
vitamin B12 injection 1 mg IM one week prior to the first dose of pemetrexed and every 3 cycles (every 9 weeks) thereafter (do not substitute oral vitamin B12 for the IM injection). After the first vitamin B12 injection, subsequent injections may be given on the same day as pemetrexed treatment.
Pemetrexed/carboplatin vs gemcitabine/carboplatin
In a multicenter, randomized phase 3 trial, no significant differences were observed for the primary end point, health-related quality of life, or the secondary end point, overall survival for patients treated with pemetrexed/carboplatin compared with gemcitabine/carboplatin (n = 436);
grade 3 or 4 toxicities were significantly worse in the gemcitabine/carboplatin arm.
Pemetrexed monotherapy
† For the treatment of advanced unresectable squamous cell head and neck cancer.
Standard Pemetrexed regimen was administered to 32 patients in a phase II trial.
Nine patients had a partial response, 15 had stable disease, and 8 progressed, which resulted in an overall response rate of 26.5% and overall survival of 6.4 months.
Pemetrexed monotherapy
† For the second line treatment of metastatic transitional-cell bladder cancer.
Standard Pemetrexed regimen
In a single-cohort phase II study, treatment with pemetrexed led to an overall response rate of 27.7% (complete response rate, 6.4%), a median overall survival time of 9.6 months, and a median time to disease progression of 2.9 months in 47 patients with stage IV transitional-cell carcinoma of the urothelium who had received 1 prior chemotherapy regimen and who had progressive disease following therapy for metastatic disease or within 12 months of neoadjuvant or adjuvant chemotherapy.
Pemetrexed monotherapy
† For the treatment of metastatic breast cancer.
Standard Pemetrexed regimen has been studied in phase II studies.
Pemetrexed monotherapy
† For the treatment of gastric cancer.
Standard Pemetrexed regimen has been studied in a phase II study of 36 patients with stage IIIB or IV disease.
Pemetrexed + gemcitabine
† For the first-line treatment of unresectable, locally advanced or metastatic pancreatic cancer.
Standard Pemetrexed regimen
At the time of review, evidence does not support the use of pemetrexed plus gemcitabine for this indication. In a phase III clinical trial, pemetrexed 500 mg/m2 IV on day 8 after gemcitabine (on days 1 and 8 of a 21-day cycle) did not improve survival end points, but did cause substantial additional toxicity.
Non-small cell lung cancer Treatment Regimens
Docetaxel-containing chemothrapy
Standard Docetaxel regimen:
Docetaxel 75 mg/m2 IV over 1 hour every 3 weeks.
Premedicate with dexamethasone 8 mg by mouth twice daily for 3 days, beginning 1 day prior to docetaxel administration, to reduce the incidence and severity of fluid retention and hypersensitivity reactions.
Docetaxel
For advanced or metastatic non-small cell lung cancer (NSCLC) after the failure of prior platinum containing chemotherapy.
Standard Docetaxel regimen. (A dose of docetaxel 100 mg/m2 in patients previously treated with chemotherapy was associated with increased hematologic toxicity, infection, and treatment-related mortality.)
In a phase II trial, the combination of docetaxel 100 mg/m2 IV over 1 hour on day 8 and gemcitabine (900 mg/m2 IV, days 1, 8) with G-CSF support, was evaluated in previously untreated patients with stage IIIB or IV NSCLC.
A partial response was seen in 37.5% of patients with an actuarial 1-year survival of 50.7%.
The combination of docetaxel (75 to 100 mg/m2 IV over 1 hour on day 1) and vinorelbine (20 to 25 mg/m2 IV on days 1 and 5) has shown response rates of 27% to 36%.
Severe neutropenia and febrile neutropenia are associated with this regimen despite colony-stimulating factor support and may limit the effectiveness of the combination.
Docetaxel + cisplatin
For first-line treatment of unresectable, locally advanced or metastatic non-small cell lung cancer (NSCLC) in combination with cisplatin in patients who have not received prior chemotherapy.
Docetaxel 75 mg/m2 IV over 1 hour immediately followed by cisplatin (75 mg/m2 IV) every 3 weeks.
Premedicate with dexamethasone 8 mg by mouth twice daily for 3 days, beginning 1 day prior to docetaxel administration, to reduce the incidence and severity of fluid retention and hypersensitivity reactions.
In a clinical trial comparing docetaxel plus cisplatin or carboplatin to a standard regimen of vinorelbine plus cisplatin,
patients in the docetaxel plus cisplatin group had a median survival time of 10.9 months vs 10 months for patients treated with vinorelbine plus cisplatin.
The overall response rates were 31.6% for docetaxel plus cisplatin vs 24.4% for vinorelbine plus cisplatin.
Additional analysis demonstrated that benefits were maintained in patients >= 65 years.
In a Japanese phase III trial, previously untreated patients with stage IV NSCLC were randomized to receive docetaxel 60 mg/m2 IV on day 1 plus cisplatin (80 mg/m2 IV day 1) every 3 to 4 weeks or vindesine (3 mg/m2 IV on days 1, 8, and 15) plus cisplatin (80 mg/m2 IV on day 1) every 4 weeks.
Of the 302 patients eligible for evaluation, patients randomized to the docetaxel arm demonstrated significant improvements as compared to those in the vindesine arm for response rates (37% vs. 21%, respectively) and median survival times (11.3 vs. 9.6 months, respectively).
In addition, quality of life measures also were favored in the docetaxel arm.
Docetaxel + carboplatin
† For first-line treatment of unresectable, locally advanced or metastatic non-small cell lung cancer (NSCLC) in combination with carboplatin.
Docetaxel 75 mg/m2 IV, followed by carboplatin (AUC 6 IV) on day 1, given every 21 days for 4 to 6 cycles.
Premedicate with dexamethasone 8 mg by mouth twice daily for 3 days, beginning 1 day prior to docetaxel administration, to reduce the incidence and severity of fluid retention and hypersensitivity reactions.
In a phase III trial of 1,203 patients with unresectable locally advanced or metastatic NSCLC,
docetaxel and platinum combinations (cisplatin or carboplatin) were compared with cisplatin/vinorelbine.
No difference was observed between docetaxel/carboplatin and cisplatin/vinorelbine in overall survival, the primary endpoint.
Grade 3 and 4 anemia and nausea/vomiting were significantly lower in both docetaxel containing arms.
In addition, hospitalizations and treatment discontinuation secondary to toxicity were higher with cisplatin/vinorelbine.
A separate phase III trial conducted in 422 patients with inoperable, locally advanced or metastatic NSCLC compared docetaxel/carboplatin to mitomycin C/cisplatin plus either ifosfamide or vinblastine.
The primary endpoint, 1-year overall survival, was not significantly different between the treatment arms.
Grade 3 and 4 neutropenia, infection, and mucositis were all significantly higher with docetaxel/carboplatin, while quality of life scores were significantly better.
Docetaxel + Gemcitabine (GD regimen)
† For first-line treatment of unresectable, locally advanced or metastatic non-small cell lung cancer (NSCLC) in combination with gemcitabine.
Multiple dosage regimens have been evaluated. Docetaxel 75, 85 or 100 mg/m2 IV on day 8 in combination with gemcitabine days 1 and 8 repeated every 21 days (GD regimen) has been studied.
Premedicate with dexamethasone 8 mg by mouth twice daily for 3 days, beginning 1 day prior to docetaxel administration, to reduce the incidence and severity of fluid retention and hypersensitivity reactions.
In a phase III trial of 311 patients with Stage IIIB or IV NSCLC, GD (docetaxel dose 85 mg/m2) was compared to cisplatin/vinorelbine (CV).
Progression-free survival (4.2 months vs. 4 months) and overall survival (11.1 months vs. 9.6 months) were not significantly different between the two arms.
Grade 3 and 4 febrile neutropenia, anemia and gastrointestinal toxicities were all significantly higher with CV, while fluid retention and grade 3 or 4 pulmonary events were greater with GD.
In another phase III trial of 413 patients comparing GD (docetaxel dose 100 mg/m2) to CV, no difference in overall survival, the primary endpoint, was observed (9 months vs. 9.7 months).
Grade 3 and 4 neutropenia and nausea/vomiting occurred more frequently with CV.
In a phase III trial of 312 patients, GD (docetaxel dose 75 mg/m2) was compared to single-agent docetaxel.
Overall survival, the primary endpoint, was significantly improved in the combination arm (9.4 months vs. 8.3 months, p = 0.037).
Cisplatin-containing chemothrapy
Cisplatin + paclitaxelel
For first-line treatment of non-small cell lung cancer (NSCLC) in patients who are not candidates for potentially curative surgery and/or radiation therapy.
Cisplatin 75 mg/m2 IV as a single dose following administration of paclitaxel every 3 weeks based on the clinical status of patient.
Cisplatin + Docetaxel
For first-line treatment of unresectable, locally advanced or metastatic non-small cell lung cancer (NSCLC) in patients who have not received prior chemotherapy.
See Docetaxel + Cisplatin in Docetaxel-containing chemothrapy above.
Cisplatin + vinorelbine
For advanced, unresectable stage III or IV non-small cell lung cancer (NSCLC)
Cisplatin 100 mg/m2 IV every 4 weeks in combination with vinorelbine (25 mg/m2 IV) once weekly.
Closely monitor CBC and renal function to determine whether a dose reduction of vinorelbine and/or cisplatin is necessary.
In the SWOG trial, most patients required a 50% dose reduction of vinorelbine at day 15 of each cycle and a 50% dose reduction of cisplatin by cycle 3.
Alternatively, cisplatin 120 mg/m2 IV on days 1 and 29, then every 6 weeks plus vinorelbine (30 mg/m2 IV) weekly may be used.
Vinorelbine in combination with cisplatin resulted in significantly improved response rates, progression-free survival and overall survival as compared to patients who received cisplatin alone for the treatment of advanced NSCLC.
Cisplatin + vinorelbine
† For adjuvant treatment of resected non-small cell lung cancer (NSCLC)
Cisplatin 100 mg/m2 on days 1, 29, 57, and 85 OR 50 mg/m2 on days 1 and 8 repeated every 4 weeks for 16 weeks has been studied in randomized, phase III studies.
Adjuvant therapy with vinorelbine (30 mg/m2 IV once weekly starting on day 1 for up to 16 doses) plus cisplatin (100 mg/m2 on days 1, 29, 57, and 85) led to significantly improved median overall survival (OS) (65.7 vs 43.7 months; hazard ratio (HR) = 0.8; 95% CI, 0.66-0.96; p = 0.017) and disease-free survival (DFS) (36.3 vs 20.7 months; HR = 0.76; 95% CI, 0.64-0.91; p = 0.002) compared with surgery +/- radiotherapy alone in a multinational, randomized, phase III study in 840 patients with completely resected stage IB-IIIA NSCLC.
The absolute OS improvement at 2- and 5-years was 4.7% and 8.4%, respectively.
In the adjuvant chemotherapy arm, grade 3 or 4 neutropenia was reported in 85% of patients and there were 7 (2%) treatment-related deaths.
In another randomized, phase III study, adjuvant therapy with vinorelbine (25 mg/m2 IV weekly for 16 weeks) plus cisplatin (50 mg/m2 on days 1 and 8 repeated every 4 weeks for 4 cycles) resulted in significantly improved median OS (94 vs 73 months; HR = 0.69; 95% CI, 0.52 to 0.91; p (adjusted) = 0.04) and relapse-free survival (not reached vs 46.7 months; HR = 0.6; 95% CI, 0.45 to 0.79; p < 0.009) compared with surgery alone in 482 patients with stage IB or II NSCLC.
The absolute improvement in 5-year OS was 11% at a median follow-up time of 9.3 years.
In a post-hoc analysis, no OS benefit was observed in patients with stage IB disease.
Grade 3 and 4 neutropenia was reported in 73% of patients and there were 2 (0.8%) treatment-related deaths.
Cisplatin + Gemcitabine (GD regimen)
For first-line treatment of inoperable, locally advanced (Stage IIIA or IIIB) or metastatic (Stage IV) non-small cell lung cancer (NSCLC)
Cisplatin 100 mg/m2 IV following gemcitabine on day 1 every 21 or 28 days. The optimal dosage schedule has not been determined.
In the clinical trials, two dosage schedules were used with similar response rates: Gemcitabine 1000 mg/m2 IV on days 1, 8, 15 of a 28-day cycle or gemcitabine 1250 mg/m2 IV on days 1 and 8 of a 21-day cycle.
Cisplatin 100 mg/m2 IV is given after the gemcitabine infusion on day 1 of either regimen. Gemcitabine doses should be adjusted for hematologic toxicity based upon the granulocyte and platelet counts taken on the day of therapy (See Dosage Adjustments).
In clinical trials, patients with inoperable, Stage IIIA or IIIB or metastatic (Stage IV) NSCLC treated with gemcitabine and cisplatin had increased overall survival and longer median time to disease progression than those patients treated with cisplatin alone or cisplatin and etoposide.
Overall response rates were higher in the gemcitabine/cisplatin group than cisplatin alone (26% vs. 10%, respectively) and cisplatin/etoposide (33% vs. 14%, respectively).
In a separate trial, gemcitabine plus cisplatin was compared to etoposide plus cisplatin in 135 patients with stage IIIB or IV NSCLC.
There was no significant difference in survival between the two treatment arms; median survival was 8.7 months for gemcitabine plus cisplatin vs. 7 months fro etoposide plus cisplatin.
The objective response rate for the gemcitabine plus cisplatin arm was 33% vs 14% on the etoposide plus cisplatin arm (Fisher's Exact p=0.01, two-sided).
Cisplatin + pemetrexed
For first-line therapy of locally advanced or metastatic non-squamous non-small cell lung cancer (NSCLC).
Cisplatin 75 mg/m2 IV on day 1 in combination with pemetrexed (500 mg/m2 IV) on day 1 repeated every 21 days for 6 cycles.
In a phase III noninferiority trial, 1725 patients with chemotherapy naive advanced NSCLC were randomized to receive treatment with either cisplatin/pemetrexed or cisplatin/gemcitabine.
No significant differences were observed in either overall survival (10.3 months vs. 10.3 months) or progression-free survival (4.8 months vs. 5.1 months).
Analysis of NSCLC histology revealed significantly worse median survival for cisplatin/pemetrexed patients with squamous-cell histology (9.4 months vs. 10.8 months).
Grade 3 or 4 neutropenia (27% vs. 15%), anemia (10% vs. 6%), and thrombocytopenia (13% vs. 4%) were all significantly higher in the cisplatin/gemcitabine arm.
Cisplatin + vinorelbine
† For adjuvant treatment of resected non-small cell lung cancer (NSCLC).
Cisplatin 100 mg/m2 on days 1, 29, 57, and 85 OR 50 mg/m2 on days 1 and 8 repeated every 4 weeks for 16 weeks has been studied in randomized, phase III studies.
Adjuvant therapy with vinorelbine (30 mg/m2 IV once weekly starting on day 1 for up to 16 doses) plus cisplatin (100 mg/m2 on days 1, 29, 57, and 85) led to significantly improved median overall survival (OS) (65.7 vs 43.7 months; hazard ratio (HR) = 0.8; 95% CI, 0.66-0.96; p = 0.017) and disease-free survival (DFS) (36.3 vs 20.7 months; HR = 0.76; 95% CI, 0.64-0.91; p = 0.002) compared with surgery +/- radiotherapy alone in a multinational, randomized, phase III study in 840 patients with completely resected stage IB-IIIA NSCLC.
The absolute OS improvement at 2- and 5-years was 4.7% and 8.4%, respectively.
In the adjuvant chemotherapy arm, grade 3 or 4 neutropenia was reported in 85% of patients and there were 7 (2%) treatment-related deaths.
In another randomized, phase III study, adjuvant therapy with vinorelbine (25 mg/m2 IV weekly for 16 weeks) plus cisplatin (50 mg/m2 on days 1 and 8 repeated every 4 weeks for 4 cycles) resulted in significantly improved median OS (94 vs 73 months; HR = 0.69; 95% CI, 0.52 to 0.91; p (adjusted) = 0.04) and relapse-free survival (not reached vs 46.7 months; HR = 0.6; 95% CI, 0.45 to 0.79; p < 0.009) compared with surgery alone in 482 patients with stage IB or II NSCLC.
The absolute improvement in 5-year OS was 11% at a median follow-up time of 9.3 years.
In a post-hoc analysis, no OS benefit was observed in patients with stage IB disease. Grade 3 and 4 neutropenia was reported in 73% of patients and there were 2 (0.8%) treatment-related deaths.
Breast Cancer Treatment Regimens
Docetaxel-based chemothrapy
Docetaxel (Taxotere) regimen
For locally advanced or metastatic breast cancer after failure of previous chemotherapy.
Docetaxel 60 to 100 mg/m2 IV over 1 hour, administered once every 3 weeks.
Premedicate with dexamethasone 8 mg by mouth twice daily for 3 days, beginning 1 day prior to docetaxel administration, to reduce the incidence and severity of fluid retention and hypersensitivity reactions.
A phase III study compared docetaxel 100 mg/m2 IV to the combination of mitomycin and vinblastine (MV) in patients with metastatic breast cancer resistant to anthracyclines.
Docetaxel produced significantly higher overall responses rates than MV (30% vs. 11.6%, respectively), median time to progression, and overall survival.
Patients who are initially dosed at 60 mg/m2 and do not experience febrile neutropenia, neutrophils less than 500 cells/mm3 for more than 1 week, severe or cumulative cutaneous reactions, or severe peripheral neuropathy may tolerate higher doses.
Docetaxel (Taxotere) regimen
† For first-line treatment of metastatic breast cancer.
Docetaxel 100 mg/m2 IV on day 1, repeated every 3 weeks until disease progression or unacceptable toxicity.
Premedicate with dexamethasone 8 mg by mouth twice daily for 3 days, beginning 1 day prior to docetaxel administration, to reduce the incidence and severity of fluid retention and hypersensitivity reactions.
In a phase III trial, 449 patients with locally advanced or metastatic breast cancer that progressed on anthracycline-containing chemotherapy, or disease that progressed during or within 12 months of anthracycline-containing adjuvant or neoadjuvant chemotherapy were randomized to receive docetaxel or paclitaxel (175 mg/m2 IV on day 1 every 3 weeks).
Overall response rate was not significantly different in the intent-to-treat population (32% vs. 25%; p = 0.10), but time-to-progression (5.7 months vs. 3.6 months; HR 1.64; p < 0.0001) and overall survival (15.4 months vs. 12.7 months; HR 1.41; p = 0.03) were significantly prolonged in the docetaxel arm.
TC regimen (Taxotere + Cyclophosphamide)
† For adjuvant treatment of operable stage I to III invasive breast cancer in combination with cyclophosphamide.
Docetaxel 75 mg/m2 IV on day 1 and cyclophosphamide 600 mg/m2 IV on day 1, given every 21 days for 4 cycles (TC regimen).
Premedicate with dexamethasone 8 mg by mouth twice daily for 3 days, beginning 1 day prior to docetaxel administration, to reduce the incidence and severity of fluid retention and hypersensitivity reactions.
In a phase III trial comparing TC to doxorubicin and cyclophosphamide (AC regimen),
the primary endpoint of 5-year disease-free survival (DFS) was significantly improved with TC (86% vs. 80%; p = 0.015).
Extended follow-up after 7 years showed a significant benefit for TC in 7-year DFS (81% vs. 75%; p = 0.033) and showed a benefit for TC in 7-year overall survival (87% vs. 82%; p = 0.032).
Edema, myalgia and arthralgia were seen more frequently with the TC regimen and nausea and vomiting were seen more frequently with the AC regimen. No formal cardiac function comparison was performed.
TAC regimen (Taxotere + Adiamycin + Cyclophosphamide)
For adjuvant treatment of operable node-positive breast cancer in combination with cyclophosphamide and doxorubicin.
Docetaxel 75 mg/m2 IV administered 1 hour after doxorubicin (50 mg/m2 IV) and cyclophosphamide (500 mg/m2 IV) every 3 weeks for 6 courses.
Premedicate with dexamethasone 8 mg by mouth twice daily for 3 days, beginning 1 day prior to docetaxel administration, to reduce the incidence and severity of fluid retention and hypersensitivity reactions.
Prophylactic colony stimulating factor support has been recommended to mitigate the risk of hematologic toxicities. Dosages should be adjusted based on toxicity.
In an open-label randomized trial, 1,491 patients (stratified based on number of positive lymph nodes) were randomized to receive either docetaxel, doxorubicin, and cyclophosphamide (TAC regimen) or doxorubicin, fluorouracil, and cyclophosphamide (FAC regimen) every 3 weeks for 6 cycles.
Results from the second interim analysis (median follow-up, 55 months) indicated that the disease-free survival was significantly longer for the TAC regimen versus the FAC regimen (HR, 0.74; 2-sided 95% CI, 0.6 to 0.92; p = 0.0047).
The primary endpoint, disease-free survival, included local and distant recurrences, contralateral breast cancer, and death from any cause.
The overall reduction in risk of relapse was 25.7% for TAC-treated patients
capecitabine
Antimetabolite antineoplastic agent; oral prodrug of 5-FU
FDA approved for the treatment of colorectal cancer and breast cancer
Has not been associated with alopecia, and myelosuppression is uncommon; hand-foot syndrome is dose-limiting
capecitabine (Xeloda) Oral Tab: 150mg, 500mg
For the treatment of metastatic breast cancer
For the treatment of metastatic breast cancer that is resistant to both paclitaxel and an anthracycline-containing chemotherapy regimen, or resistant to paclitaxel and for whom further anthracycline therapy is not indicated (e.g., patients who have received cumulative doses of 400 mg/m2 of doxorubicin or doxorubicin equivalent), as monotherapy.
NOTE: Resistance is defined as progressive disease while on treatment regardless of an initial response or relapse within 6 months of treatment completion with an anthracycline-containing adjuvant regimen.
Oral dosage
1,250 mg/m2 by mouth twice daily (approximately 12 hours apart), within 30 minutes after a meal on days 1 to 14, followed by 1 week of rest. Repeat every 3 weeks.
Dosages should be adjusted based on toxicity during treatment cycle; once a dosage is reduced, it should not be increased at a later time.
In a subset of patients with stage IV breast cancer that was resistant to both paclitaxel and an anthracycline (n = 43) from an open-label,
single-arm clinical trial, treatment with capecitabine resulted in a partial response rate of 25.6% (95% CI, 13.5% to 41.2%) for a median duration of 5 months (range, 2.1 months to 7.6 months).
The median time to progression in these patients was 3.3 months and the median survival was 8.4 months.
capecitabine + docetaxel
For the treatment of metastatic breast cancer after failure of prior anthracycline-containing chemotherapy
Oral dosage
Capecitabine 1,250 mg/m2 by mouth twice daily (approximately 12 hours apart), within 30 minutes after a meal on days 1 to 14, followed by 1 week of rest.
Docetaxel 75 mg/m2 IV over 1 hour on day 1. Repeat every 3 weeks. Premedicate with dexamethasone.
Dosages should be adjusted based on toxicity during treatment cycle; once a dosage is reduced, it should not be increased at a later time.
In a multicenter, randomized, open-label, phase III trial of 511 patients randomized to docetaxel/capecitabine or docetaxel alone, overall response rate (32% vs. 23%, p = 0.009), time-to-progression (6.1 months vs. 4.2 months, HR 0.643, p = 0.0001), and overall survival (14.5 months vs. 11.5 months, HR 0.775, p = 0.0126) were improved in the combination arm.
Docetaxel/capecitabine patients experienced more hand-foot syndrome, diarrhea, stomatitis, nausea, and vomiting. Neutropenic fever, myalgia, arthralgia, and fatigue occurred more frequently with single-agent docetaxel.
br>
Docetaxel-based chemothrapy with concomitant Trastuzumab
Docetaxel + Trastuzumab
† For HER2-positive metastatic breast cancer in combination with trastuzumab
(NOTE: Trastuzumab is FDA approved for use in combination with docetaxel for the treatment of metastatic HER-2 positive breast cancer)
Docetaxel 100 mg/m2 IV on day 1repeated every 21 days for 6 to 8 cycles,
in combination with trastuzumab (4 mg/kg IV on day 1 of first cycle only, then 2 mg/kg IV weekly).
Premedicate with dexamethasone 8 mg by mouth twice daily for 3 days, beginning 1 day prior to docetaxel administration, to reduce the incidence and severity of fluid retention and hypersensitivity reactions.
Trastuzumab should be continued until disease progression or unacceptable toxicity.
Docetaxel in combination with trastuzumab was studied in a phase II trial of 186 patients with previously untreated HER2-positive metastatic breast cancer.
In this trial, docetaxel/trastuzumab was compared to docetaxel alone. Overall response rate (ORR) (61% vs. 34%; p = 0.0002),
the primary endpoint, time-to-progression (TTP) (11.7 months vs. 6.1 months; p = 0.0001), and
overall survival (OS) (31.2 months vs. 22.7 months; p = 0.0325) were all significantly improved in the docetaxel/trastuzumab arm.
This benefit has been confirmed in other clinical trials, including a phase III trial of 263 patients where it was shown to produce a similar TTP and OS to docetaxel/carboplatin/trastuzumab, a regimen with known activity against breast cancer in the adjuvant setting.
In a phase II trial of 30 women with HER2-overexpressed metastatic breast cancer, docetaxel 35 mg/m2 IV on days 1, 8, and 15 in combination with trastuzumab on days 1, 8, and 15, every 28 days was studied, and treatment was continued until disease progression or unacceptable toxicity; ORR was 63%.
Trastuzumab + Docetaxel
† For the treatment of HER2-positive metastatic breast cancer, in combination with docetaxel.
Trastuzumab 4 mg/kg IV over 90 minutes on day 1 (of first cycle only), then 2 mg/kg IV over 30 minutes once weekly until disease progression or unacceptable toxicity.
In combination with docetaxel (100 mg/m2 IV on day 1) every 21 days for 6 to 8 cycles.
In a phase 2 trial of 186 patients with previously untreated HER2-positive metastatic breast cancer,
docetaxel/trastuzumab significantly improved overall response rate (ORR) compared to docetaxel alone (61% vs 34%);
time-to-progression (TTP) (11.7 vs. 6.1 months) and
overall survival (OS) (31.2 vs. 22.7 months) were also significantly improved in the docetaxel/trastuzumab arm.
This benefit has been confirmed in other clinical trials, including a phase 3 trial of 263 patients where it was shown to produce a similar TTP and OS to docetaxel/carboplatin/trastuzumab, a regimen with known activity against breast cancer in the adjuvant setting.
In a phase 2 trial of 30 women with HER2-overexpressed metastatic breast cancer, docetaxel 35 mg/m2 IV on days 1, 8, and 15 was studied in combination with trastuzumab 2 mg/kg IV (4 mg/kg IV on day 0 of first cycle only) on days 1, 8, and 15, repeated every 28 days until disease progression or unacceptable toxicity; the ORR was 63%.
Trastuzumab + TAC regimen
For adjuvant treatment of HER2-positive, node-positive or node-negative (ER/PR negative or with one high-risk feature) breast cancer in combination with docetaxel, after completion of doxorubicin and cyclophosphamide chemotherapy (AC-TH).
Trastuzumab 4 mg/kg IV over 90 minutes on day 1 (of first cycle only), then 2 mg/kg IV over 30 minutes once weekly for a total of 12 weeks,
in combination with docetaxel (100 mg/m2 IV every 21 days) beginning on day 1 for a total of 4 cycles (12 weeks).
On week 13, begin trastuzumab 6 mg/kg IV over 30 to 90 minutes every 3 weeks as monotherapy for a total of 52 weeks of trastuzumab therapy.
Begin docetaxel plus trastuzumab after the completion of 4 cycles of AC chemotherapy (doxorubicin 60 mg/m2 IV and cyclophosphamide 600 mg/m2 IV every 21 days);
trastuzumab should NOT be administered concurrently with doxorubicin and cyclophosphamide.
Extending treatment beyond 1 year is not recommended as it increased the risk of asymptomatic cardiac dysfunction (2 years, 8.1%; 1 year, 4.6%) and grade 3 or higher adverse reactions (2 years, 20.4%; 1 year, 16.3%) without improving efficacy.
In a randomized clinical trial of patients with HER2-positive breast cancer, adjuvant treatment with docetaxel plus trastuzumab after completion of AC chemotherapy significantly improved disease free survival compared with docetaxel alone.
Docetaxel (Taxotere) + carboplatin + Trastuzumab (Herceptin) - TCH regimen
† For adjuvant treatment of HER2 overexpressing, node-positive or node negative (ER/PR negative or with one high-risk feature) breast cancer, in combination with trastuzumab and carboplatin (TCH regimen).
(NOTE: Trastuzumab is FDA approved for the adjuvant treatment of HER2 overexpressing, node-positive or node negative (ER/PR negative or with one high-risk feature) breast cancer, in combination with docetaxel and carboplatin.)
Docetaxel 75 mg/m2 IV on day 1 with carboplatin (AUC 6 IV on day 1), repeated every 3 weeks for a total of 6 cycles,
given concurrently with trastuzumab (4 mg/kg IV on week 1, then 2 mg/kg IV weekly for 18 total weekly doses, then 6 mg/kg IV every 3 weeks for the balance of 52 weeks total)(TCH regimen).
Premedicate with dexamethasone 8 mg by mouth twice daily for 3 days, beginning 1 day prior to docetaxel administration, to reduce the incidence and severity of fluid retention and hypersensitivity reactions.
In a phase III trial of 3,222 patients with node-positive or high-risk node-negative breast cancer, patients were randomized to receive TCH, doxorubicin/cyclophosphamide followed by docetaxel/trastuzumab (AC-TH), or the control arm, doxorubicin/cyclophosphamide followed by docetaxel (AC-T).
At a median follow-up of 65 months, 5-year disease-free survival (81% vs. 75%; p = 0.04) and 5-year overall survival (91% vs. 87%; p =0.038) were significantly improved by TCH compared to AC-T.
Screening, Immunization, and Treatment for Viral Diseases in Patients with Cancer
Screening for Hepatitis B Virus Before Starting Cancer Treatment
ASCO has updated a provisional clinical opinion recommending that patients with risk factors for hepatitis B virus (HBV) infection or those who will be receiving cancer treatment likely to cause a reactivation of HBV be screened for HBV before beginning cancer treatment. A provisional clinical opinion offers direction to doctors and others who treat people with cancer after the publication or presentation of potentially practice-changing information.
About hepatitis B
HBV is a virus that infects the liver and is detected with a blood test. Long-term infection with HBV damages the liver and can lead to liver cancer, scarring of the liver called cirrhosis, liver failure, and even death. HBV is spread by coming into contact with body fluids, such as blood or semen, of a person who is infected.
HBV infection can be prevented with a three-shot vaccine series. Not all people with HBV infection need treatment for the infection, depending on the results of screening. Some may need regular testing to watch for a reactivation. Others may need treatment with drugs called antiviral medications to avoid reactivation.
Those at high risk for hepatitis B infection include:
People who use injection drugs
People with multiple sex partners or those diagnosed with a sexually transmitted disease
Men who have sex with men
People who have sexual contact with or live in the same household as someone who is already infected with HBV
Infants who are born to mothers infected with HBV
Infants and children of immigrants from areas with high rates of HBV infection, such as Eastern Europe, Asia, Africa, the Middle East, and the Pacific Islands
Health care workers or other employees who come into contact with human blood
Patients who are receiving hemodialysis for kidney disease
HBV and cancer
For people with cancer and HBV infection who need cancer treatment that lowers the immune system, there is a risk that an inactive, or latent, infection with hepatitis B can become active again. This is called reactivation, and it could lead to serious liver problems and complicate cancer treatment.
It is not exactly known how a latent HBV infection becomes active or causes problems for people with specific cancers or specific treatments. It is also not clear why some cancer treatments cause reactivation while others do not. Treatments that lower the immune system and have a high chance of leading to reactivation include the following:
High-dose chemotherapy for a stem cell/bone marrow transplantation
Treatments that lower the number of B cells, such as anti-CD20 therapy. These include rituximab (Rituxan) and ofatumumab (Azerra), which may be used to treat cancers of the blood, bone marrow, lymph, or lymphatic system.
Recommendations for HBV screening and treatment
In the updated provisional clinical opinion, ASCO recommends the following:
Right now, there is not enough scientific evidence to recommend screening every patient for HBV.
Patients should receive HBV screening if they have a high risk of HBV infection or are going to receive a cancer treatment that increases the risk of HBV reactivation. These types of treatments include anti-CD20 therapy or stem cell/bone marrow transplantation.
Testing should include two tests for HBV. One test looks for the hepatitis B surface antigen (HBsAg). The other test looks for the hepatitis B core antibody (anti-HBc). HBV reactivation can happen if both tests are positive or if only the anti-HBc test is positive.
For patients who test positive for both HBV tests, antiviral therapy should be given before or along with cancer treatment.
Those who test positive only for the anti-HBc test can be considered for antiviral therapy or be monitored for reactivation of HBV with blood tests every three months instead of antiviral therapy.
Some cancer centers follow what is called universal HBV screening, which means that all patients are screened for HBV before cancer treatment begins. Currently, there is no strong evidence to support either starting or stopping universal screening. Until more research of this topic is available, HBV screening is recommended at least for patients with HBV risk factors and/or patients who are about to receive a cancer treatment linked with a high risk of HBV reactivation.
What this means for patients
Talk with your doctor about whether you need HBV testing given your health, risk factors for HBV infection, and cancer treatment plan. It is important to tell your health care team if you fit the criteria for a high risk of HBV infection and if you have been diagnosed with HBV in the past. Having an HBV test and receiving antiviral therapy if you test positive can help you avoid HBV reactivation.
Patients who need antiviral medication may need to take it during cancer treatment and for six months to one year after cancer treatment. This additional treatment could add to the overall cost of treatment. In addition, patients receiving antiviral therapy may need to visit a hepatitis specialist. If you have a high risk of HBV and/or are going to receive cancer treatment that could reactivate the virus, talk with your doctor about HBV screening and any concerns you may have about managing your care.
Immunization
Shingles vaccine
1. Zostavax (Merck)
2. Shingrix (GSK's 2 dose schedule)
• CDC recommends that healthy adults 50 years and older get two doses of Shingrix, 2 to 6 months apart. Shingrix provides strong protection against shingles and postherpetic neuralgia (PHN). Shingrix is the preferred vaccine, over Zostavax, a shingles vaccine in use since 2006.
Treatment of Postherpetic neuralgia
Lidocaine skin patches (Lidocaine Transdermal Patch) - Lidoderm®
Lidocaine patches are used to relieve the pain of post-herpetic neuralgia.
It is applied only once a day as needed for pain. Never apply more than three patches at one time, and never wear patches for more than 12 hours per day. Using too many patches or leaving patches on for too long may cause serious side effects.
It should be applied only to intact skin.
To apply the patches, follow these steps:
Look at the skin that you plan to cover with a lidocaine patch. If the skin is broken or blistered, do not apply a patch to that area.
Use scissors to remove the outer seal from the package. Then pull apart the zipper seal.
Remove up to three patches from the package and press the zipper seal tightly together. The remaining patches may dry out if the zipper seal is not tightly closed.
Cut patch(es) to the size and shape that will cover your most painful area.
Peel the transparent liner off the back of the patch(es).
Press the patch(es) firmly onto your skin. If you are applying a patch to your face, be careful not to let it touch your eyes. If you do get lidocaine in your eye, wash it with plenty of water or saline solution.
Wash your hands after handling lidocaine patches.
Do not reuse lidocaine patches. After you are finished using a patch, remove it and dispose of it out of reach of children and pets. Used patches contain enough medication to seriously harm a child or pet.
Before using lidocaine patches, tell your doctor and pharmacist if you are allergic to lidocaine; other local anesthetics such as bupivacaine (Marcaine), etidocaine (Duranest), mepivacaine (Carbocaine, Prolocaine), or prilocaine (Citanest); or any other medications.
LIDODERM (lidocaine patch 5%), Need Prescription.
Each adhesive patch contains 700 mg of lidocaine (50 mg per gram adhesive) in an aqueous base.
LIDODERM (lidocaine patch 4% or less), Lidocaine3.6%,Menthol1.25%, Over-The-Counter.
DOSAGE AND ADMINISTRATION
Apply LIDODERM to intact skin to cover the most painful area. Apply the prescribed number of patches (maximum of 3), only once for up to 12 hours within a 24 hour period. Patches may be cut into smaller sizes with scissors prior to removal of the release liner. (See Handling And Disposal) Clothing may be worn over the area of application. Smaller areas of treatment are recommended in a debilitated patient, or a patient with impaired elimination.
If irritation or a burning sensation occurs during application, remove the patch (es) and do not reapply until the irritation subsides.
When LIDODERM is used concomitantly with other products containing local anesthetic agents, the amount absorbed from all formulations must be considered.
LIDODERM may not stick if it gets wet. Avoid contact with water, such as bathing, swimming or showering.
HOW SUPPLIED
LIDODERM (lidocaine patch 5%) is available as the following:
Carton of 30 patches, packaged into individual child-resistant envelopes
Store at 25°C (77°F); excursions permitted to 15°-30°C (59°-86°F). [See USP Controlled Room Temperature].
Prices and coupons for 30 patches 5% of lidocaine, $300, Discount $80-150.
Ref: Arthritis Foundation: Supplement Guide: Capsaicin https://www.arthritis.org/living-with-arthritis/treatments/natural/supplements-herbs/guide/capsaicin.php
Capsaicin (Capsicum frutescens)
Origin: The highly purified, heat-producing component in chili peppers
What we know: Applied as a topical cream, gel or patch, capsaicin activates specific nerve receptors causing local heat, stinging and/or itching sensations. Prolonged activation of these receptors causes them to lose their ability to function properly (and process pain signals) for extended periods of time. Capsaicin must be used regularly to keep the nerve receptors from working properly and processing pain signals.
Studies: Many studies have shown that capsaicin effectively reduces pain from osteoarthritis, rheumatoid arthritis and fibromyalgia. In a 2010 German study, joint pain decreased nearly 50 percent after three weeks' use of 0.05 percent capsaicin cream.
Dosage: Most capsaicin products – such as Zostrix, Zostrix HP, Capzasin-P and others – contain between 0.025 to 0.075 percent concentrations. Apply regularly three times daily.