Non-Hodgkin Lymphoma (NHL)
Lymphoma is a cancer of the lymphatic system. Lymphoma begins when healthy B cells, T cells, or NK cells in the lymphatic system change and grow out of control, which may form a tumor. Hodgkin lymphoma is a specific type of lymphoma. Non-Hodgkin lymphoma (NHL) is a term that refers to a group of cancers of the lymphatic system. These cancers can have different symptoms and signs, findings on a physical examination, and treatments.
Because lymphatic tissue is found in most parts of the body, NHL can start almost anywhere and can spread, or metastasize, to almost any organ. It often begins in the lymph nodes, liver, spleen, or bone marrow. However, it can also involve the stomach, intestines, skin, thyroid gland, brain, or any other part of the body.
(ASCO)
The non-Hodgkin lymphomas (NHL) are a heterogeneous group of lymphoproliferative malignancies with differing patterns of behavior and responses to treatment.
Like Hodgkin lymphoma, NHL usually originates in lymphoid tissues and can spread to other organs. NHL, however, is much less predictable than Hodgkin lymphoma and has a far greater predilection to disseminate to extranodal sites. The prognosis depends on the histologic type, stage, and treatment.
Incidence and Mortality
Estimated new cases and deaths from NHL in the United States in 2018:
• New cases: 74,680. • Deaths: 19,910.
Prognosis and Survival
NHL can be divided into two prognostic groups: the indolent lymphomas and the aggressive lymphomas.
Indolent NHL types have a relatively good prognosis with a median survival as long as 20 years, but they usually are not curable in advanced clinical stages. Early-stage (stage I and stage II) indolent NHL can be effectively treated with radiation therapy alone. Most of the indolent types are nodular (or follicular) in morphology.
The aggressive type of NHL has a shorter natural history, but a significant number of these patients can be cured with intensive combination chemotherapy regimens.
In general, with modern treatment of patients with NHL, overall survival at 5 years is over 60%.
Of patients with aggressive NHL, more than 50% can be cured.
The vast majority of relapses occur in the first 2 years after therapy.
The risk of late relapse is higher in patients who manifest both indolent and aggressive histologies.
While indolent NHL is responsive to immunotherapy, radiation therapy, and chemotherapy, a continuous rate of relapse is usually seen in advanced stages.
Patients, however, can often be re-treated with considerable success as long as the disease histology remains low grade.
Patients who present with or convert to aggressive forms of NHL may have sustained complete remissions with combination chemotherapy regimens or aggressive consolidation with marrow or stem cell support.
Late Effects of Treatment for Adult NHL
Late effects of treatment for non-Hodgkin lymphoma (NHL) have been observed.
Pelvic radiation therapy and large cumulative doses of cyclophosphamide have been associated with a high risk of permanent sterility.
For as many as three decades after diagnosis, patients are at a significantly elevated risk for second primary cancers, especially the following:
• Lung cancer.
• Brain cancer.
• Kidney cancer.
• Bladder cancer.
• Melanoma.
• Hodgkin lymphoma.
• Acute nonlymphocytic leukemia.
Left ventricular dysfunction was a significant late effect in long-term survivors of high-grade NHL who received more than 200 mg/m2 of doxorubicin.
Myelodysplastic syndrome and acute myelogenous leukemia are late complications of myeloablative therapy with autologous bone marrow or peripheral blood stem cell support, as well as conventional chemotherapy-containing alkylating agents.
Most of these patients show clonal hematopoiesis even before the transplantation, suggesting that the hematologic injury usually occurs during induction or reinduction chemotherapy.
With a median 10-year follow-up after autologous bone marrow transplantation (BMT) with conditioning using cyclophosphamide and total-body radiation therapy, in a series of 605 patients, the incidence of a second malignancy was 21%, and 10% of those were solid tumors.
Successful pregnancies with children born free of congenital abnormalities have been reported in young women after autologous BMT.
Some patients have osteopenia or osteoporosis at the start of therapy; bone density may worsen after therapy for lymphoma.
Risk Factors
The exact cause of NHL is not known, and most people diagnosed with NHL will not learn what caused it. However, the following factors may raise a person’s risk of developing NHL:
• Age. The risk of NHL increases with age. The most common subtypes occur most often in people in their 60s and 70s.
• Gender. Men are very slightly more likely to develop NHL than women.
• Bacterial infections. Some types of NHL are associated with specific infections. For example, mucosa-associated lymphoid tissue (MALT) lymphoma of the stomach is thought to be caused by an infection with bacteria called Helicobacter pylori. If this lymphoma is diagnosed very early, it will sometimes go away if the infection is cured with antibiotics. Infections may also cause other types of MALT lymphoma, including those affecting the lungs, tear glands, and skin.
• Viruses. Viruses cause some types of NHL. For example, as explained in the Subtypes section, Epstein-Barr virus (EBV, which is the virus that causes mononucleosis, also known as "mono") is associated with some types of NHL. Those include Burkitt lymphoma, lymphomas occurring after an organ transplant, and, rarely, other lymphomas in people who are otherwise healthy. However, the virus is probably not the only factor that determines cancer risk, so people who have had mononucleosis do not necessarily have an increased risk of developing NHL in the future. In addition, hepatitis C infection has been associated with an increased risk of marginal zone lymphomas of the spleen. Researchers have also found other viruses to be important in causing other, rare types of lymphoma.
• Immune deficiency disorders. Immune system disorders, such as HIV/AIDS, increase the risk of NHL, especially the aggressive B-cell lymphomas.
• Autoimmune disorders. People with autoimmune disorders, such as rheumatoid arthritis and Sjögren syndrome, have an increased risk of developing certain types of NHL. Also, some drugs used to treat autoimmune disorders may increase the risk of NHL.
• Organ transplantation. Organ transplant recipients have a higher risk of NHL. This is because of the drugs patients must take to reduce immune system function in order to protect the transplanted organ from rejection.
• Previous cancer treatment. Previous treatment with certain drugs for other types of cancer may increase the risk of NHL.
• Chemical exposure. Exposure to chemicals, such as certain pesticides, herbicides and petrochemicals, may increase the risk of NHL.
• Genetic factors. Early studies have found several genetic changes that may be associated with a small number of lymphoma cases. Currently there are no widely accepted genetic tests to identify inherited risk factors for NHL or that reliably predict a person’s risk of developing NHL.
• Vaccines. A number of studies have found an association between Bacillus Calmette–Guerin (BCG) vaccination and an increased risk of NHL. BCG is a vaccine for tuberculosis disease. Conversely, research has also associated other vaccinations with a decreased risk of NHL, such as those for smallpox, cholera, yellow fever, influenza, measles, tetanus, and polio. Overall, the relationship between vaccinations and lymphoma remains unclear and controversial.
• Breast implants. Having breast implants can increase the risk of breast lymphomas.
• Exposure to ionizing radiation. This can include exposure to radiation from atomic bombs, nuclear reactor accidents, and medical radiation therapy.
• Dietary risk factors. There is some inconclusive evidence that being overweight or having a diet filled with fatty foods or red meat may slightly increase the risk of lymphoma.
Classification of Adult NHL
Cellular Classification of Adult NHL
A pathologist should be consulted before a biopsy because some studies require special preparation of tissue (e.g., frozen tissue).
Knowledge of cell surface markers and immunoglobulin and T-cell receptor gene rearrangements may help with diagnostic and therapeutic decisions.
The clonal excess of light-chain immunoglobulin may differentiate malignant from reactive cells. Since the prognosis and the approach to treatment are influenced by histopathology, outside biopsy specimens should be carefully reviewed by a hematopathologist who is experienced in diagnosing lymphomas.
Although lymph node biopsies are recommended whenever possible, sometimes immunophenotypic data are sufficient to allow diagnosis of lymphoma when fine-needle aspiration cytology is preferred.
Historical Classification Systems
Historically, uniform treatment of patients with non-Hodgkin lymphoma (NHL) has been hampered by the lack of a uniform classification system.
In 1982, results of a consensus study were published as the Working Formulation. The Working Formulation combined results from six major classification systems into one classification.
This allowed comparison of studies from different institutions and countries. The Rappaport classification, which also follows, is no longer in common use.
Historical Classification Systems for Non-Hodgkin Lymphoma (NHL)
| Working Formulation | Rappaport Classification |
| Low grade | |
| A. Small lymphocytic, consistent with chronic lymphocytic leukemia | Diffuse lymphocytic, well-differentiated |
| B. Follicular, predominantly small-cleaved cell | Nodular lymphocytic, poorly differentiated |
| C. Follicular, mixed small-cleaved, and large cell | Nodular mixed, lymphocytic, and histiocytic |
| Intermediate grade | |
| D. Follicular, predominantly large cell | Nodular histiocytic |
| E. Diffuse, small-cleaved cell | Diffuse lymphocytic, poorly differentiated |
| F. Diffuse mixed, small and large cell | Diffuse mixed, lymphocytic, and histiocytic |
| G. Diffuse, large cell, cleaved, or noncleaved cell | Diffuse histiocytic |
| High grade | |
| H. Immunoblastic, large cell | Diffuse histiocytic |
| I. Lymphoblastic, convoluted, or nonconvoluted cell | Diffuse lymphoblastic |
| J. Small noncleaved-cell, Burkitt, or non-Burkitt | Diffuse undifferentiated Burkitt or non-Burkitt |
Current Classification Systems
As the understanding of NHL has improved and as the histopathologic diagnosis of NHL has become more sophisticated with the use of immunologic and genetic techniques, a number of new pathologic entities have been described.
In addition, the understanding and treatment of many of the previously described pathologic subtypes have changed. As a result, the Working Formulation has become outdated and less useful to clinicians and pathologists.
Thus, European and American pathologists have proposed a new classification, the Revised European American Lymphoma (REAL) classification. Since 1995, members of the European and American Hematopathology societies have been collaborating on a new World Health Organization (WHO) classification, which represents an updated version of the REAL system.
The WHO modification of the REAL classification recognizes three major categories of lymphoid malignancies based on morphology and cell lineage:
B-cell neoplasms, T-cell/natural killer (NK)-cell neoplasms, and Hodgkin lymphoma (HL).
Both lymphomas and lymphoid leukemias are included in this classification because both solid and circulating phases are present in many lymphoid neoplasms and distinction between them is artificial.
For example, B-cell chronic lymphocytic leukemia (CLL) and B-cell small lymphocytic lymphoma are simply different manifestations of the same neoplasm, as are lymphoblastic lymphomas and acute lymphocytic leukemias.
Within the B-cell and T-cell categories, two subdivisions are recognized: precursor neoplasms, which correspond to the earliest stages of differentiation, and more mature differentiated neoplasms.
Molecular Diagnosis
Waldenstrom’s Macroglobulinaemia : Expression of CD19, CD20, CD22 and CD79aon the lymphocytic as well as CD38 on the plasmacytic component (small variations can occur)
Molecular Diagnosis
=====================
Updated REAL/WHO classification
B-cell neoplasms
1. Precursor B-cell neoplasm: precursor B-acute lymphoblastic leukemia/lymphoblastic lymphoma (LBL).
2. Peripheral B-cell neoplasms.
a. B-cell CLL/small lymphocytic lymphoma.
b. B-cell prolymphocytic leukemia.
c. Lymphoplasmacytic lymphoma/immunocytoma.
d. Mantle cell lymphoma.
e. Follicular lymphoma.
f. Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphatic tissue (MALT) type.
g. Nodal marginal zone B-cell lymphoma (± monocytoid B-cells).
h. Splenic marginal zone lymphoma (± villous lymphocytes).
i. Hairy cell leukemia.
j. Plasmacytoma/plasma cell myeloma.
k. Diffuse large B-cell lymphoma.
l. Burkitt lymphoma.
T-cell and putative NK-cell neoplasms
1. Precursor T-cell neoplasm: precursor T-acute lymphoblastic leukemia/LBL.
2. Peripheral T-cell and NK-cell neoplasms.
a. T-cell CLL/prolymphocytic leukemia.
b. T-cell granular lymphocytic leukemia.
c. Mycosis fungoides (including Sézary syndrome).
d. Peripheral T-cell lymphoma, not otherwise characterized.
e. Hepatosplenic gamma/delta T-cell lymphoma.
f. Subcutaneous panniculitis-like T-cell lymphoma.
g. Angioimmunoblastic T-cell lymphoma.
h. Extranodal T-/NK-cell lymphoma, nasal type.
i. Enteropathy-type intestinal T-cell lymphoma.
j. Adult T-cell lymphoma/leukemia (human T-lymphotrophic virus [HTLV] 1+).
k. Anaplastic large cell lymphoma, primary systemic type.
l. Anaplastic large cell lymphoma, primary cutaneous type.
m. Aggressive NK-cell leukemia.
Hodgkin Lymphoma (HL)
1. Nodular lymphocyte-predominant HL.
2. Classical HL.
a. Nodular sclerosis HL.
b. Lymphocyte-rich classical HL.
c. Mixed-cellularity HL.
d. Lymphocyte-depleted HL.
The REAL classification encompasses all the lymphoproliferative neoplasms.
Refer to the following PDQ summaries for more information:
• Adult Acute Lymphoblastic Leukemia Treatment
• Adult Hodgkin Lymphoma Treatment
• AIDS-Related Lymphoma Treatment
• Chronic Lymphocytic Leukemia Treatment
• Hairy Cell Leukemia Treatment
• Mycosis Fungoides (Including Sézary Syndrome) Treatment
• Plasma Cell Neoplasms (Including Multiple Myeloma) Treatment
• Primary CNS Lymphoma Treatment
PDQ modification of REAL classification of lymphoproliferative diseases
1. Plasma cell disorders.
a. Bone.
b. Extramedullary.
i. Monoclonal gammopathy of undetermined significance.
ii. Plasmacytoma.
iii. Multiple myeloma.
iv. Amyloidosis.
2. Hodgkin Lymphoma (HL)
a. Nodular sclerosis HL.
b. Lymphocyte-rich classical HL.
c. Mixed-cellularity HL.
d. Lymphocyte-depleted HL.
3. Indolent lymphoma/leukemia.
a. Follicular lymphoma (follicular small-cleaved cell [grade 1], follicular mixed small-cleaved, and large cell [grade 2], and diffuse, small-cleaved cell).
b. Chronic lymphocytic leukemia/small lymphocytic lymphoma.
c. Lymphoplasmacytic lymphoma (Waldenström macroglobulinemia).
d. Extranodal marginal zone B-cell lymphoma (MALT lymphoma).
e. Nodal marginal zone B-cell lymphoma (monocytoid B-cell lymphoma).
f. Splenic marginal zone lymphoma (splenic lymphoma with villous lymphocytes).
g. Hairy cell leukemia.
h. Mycosis fungoides (including Sézary syndrome).
i. T-cell granular lymphocytic leukemia.
j. Primary cutaneous anaplastic large cell lymphoma/lymphomatoid papulosis (CD30+).
k. Nodular lymphocyte–predominant Hodgkin lymphoma.
4. Aggressive lymphoma/leukemia.
a. Diffuse large cell lymphoma (includes diffuse mixed-cell, diffuse large cell, immunoblastic, and T-cell rich large B-cell lymphoma).
Distinguish:
i. Mediastinal large B-cell lymphoma.
ii. Follicular large cell lymphoma (grade 3).
iii. Anaplastic large cell lymphoma (CD30+).
iv. Extranodal NK-/T-cell lymphoma, nasal type/aggressive NK-cell leukemia/blastic NK-cell lymphoma.
v. Lymphomatoid granulomatosis (angiocentric pulmonary B-cell lymphoma).
vi. Angioimmunoblastic T-cell lymphoma.
vii. Peripheral T-cell lymphoma, unspecified.
• Subcutaneous panniculitis-like T-cell lymphoma.
• Hepatosplenic T-cell lymphoma.
viii. Enteropathy-type T-cell lymphoma.
ix. Intravascular large B-cell lymphoma.
b. Burkitt lymphoma/Burkitt cell leukemia/Burkitt-like lymphoma.
c. Precursor B-cell or T-cell lymphoblastic lymphoma/leukemia.
d. Primary central nervous system (CNS) lymphoma. (Refer to the PDQ summary on Primary CNS Lymphoma Treatment.)
e. Adult T-cell leukemia/lymphoma (HTLV 1+).
f. Mantle cell lymphoma.
g. Posttransplantation lymphoproliferative disorder.
h. AIDS-related lymphoma.
i. True histiocytic lymphoma.
j. Primary effusion lymphoma.
k. B-cell or T-cell prolymphocytic leukemia.
l. Plasmablastic lymphoma.
Clinical-Pathological Types
Indolent NHL and Aggressive NHL
Indolent NHL
Indolent non-Hodgkin lymphoma (NHL) includes the following subtypes:
• Follicular lymphoma.
• Lymphoplasmacytic lymphoma (Waldenström macroglobulinemia).
• Marginal zone lymphoma.
• Splenic marginal zone lymphoma.
• Primary cutaneous anaplastic large cell lymphoma.
Follicular lymphoma
Follicular lymphoma comprises 20% of all NHL and as many as 70% of the indolent lymphomas reported in American and European clinical trials.
Most patients with follicular lymphoma are age 50 years and older and present with widespread disease at diagnosis.
Nodal involvement is most common and is often accompanied by splenic and bone marrow disease.
Rearrangement of the bcl-2 gene is present in more than 90% of patients with follicular lymphoma; overexpression of the bcl-2 protein is associated with the inability to eradicate the lymphoma by inhibiting apoptosis.
Prognosis
Despite the advanced stage, the median survival ranges from 8 to 15 years, leading to the designation of being indolent.
Patients with advanced-stage follicular lymphoma are not cured with current therapeutic options. The rate of relapse is fairly consistent over time, even in patients who have achieved complete responses to treatment.
Watchful waiting, i.e., the deferring of treatment until the patient becomes symptomatic, is an option for patients with advanced-stage follicular lymphoma.
An international index for follicular lymphoma (i.e., the Follicular Lymphoma International Prognostic Index [FLIPI]) identified five significant risk factors prognostic of overall survival (OS):
1. Age (≤60 years vs. >60 years).
2. Serum lactate dehydrogenase (LDH) (normal vs. elevated).
3. Stage (stage I or stage II vs. stage III or stage IV).
4. Hemoglobin level (≥120 g/L vs. <120 g/L).
5. Number of nodal areas (≤4 vs. >4).
Patients with none or one risk factor have an 85% 10-year survival rate, while three or more risk factors confer a 40% 10-year survival rate.
In a revised FLIPI-2, an elevated beta-2-microglobulin and lymph node size of more than 6 cm are proposed prognostic factors instead of serum LDH and the number of nodal areas.
Although the FLIPI and FLIPI-2 indices can predict progression-free survival (PFS) and OS, the scores cannot be used to establish the need for therapy, nor can they be used to predict response to therapy. The primary use of FLIPI or FLIPI-2 is to assure a balance of prognostic factors or to define entry requirements in randomized clinical trials.
Individuals with an adverse FLIPI score may well benefit from watchful waiting or may still respond well to initial therapy. Gene expression profiles of tumor biopsy specimens suggest that follicular lymphoma that is surrounded by infiltrating T-lymphocytes has a much longer median survival (13.6 years) than follicular lymphoma that is surrounded by dendritic and monocytic cells (3.9 years) (P < .001).
Two retrospective analyses identified a high-risk group that had a 50% OS rate at 5 years when relapse occurred after induction chemoimmunotherapy at 24 or 30 months; this has not been validated in prospective studies or an independent cohort. These higher-risk patients represent a target population for clinical trials.
Follicular, small-cleaved cell lymphoma and follicular mixed small-cleaved and large cell lymphoma do not have reproducibly different disease-free survival or OS.
Therapeutic approaches
Therapeutic options include:
• watchful waiting;
• rituximab, an anti-CD20 monoclonal antibody, alone or with purine nucleoside analogs;
• oral alkylating agents; and
• combination chemotherapy.
• Radiolabeled monoclonal antibodies, vaccines, and autologous or allogeneic bone marrow or peripheral stem cell transplantation are also under clinical evaluation.
Currently, no randomized trials have mature results to guide clinicians about the initial choice of rituximab, nucleoside analogs, alkylating agents, combination chemotherapy, radiolabeled monoclonal antibodies, or combinations of these options.
On a comparative basis, it is difficult to prove benefit when relapsing disease is followed with watchful waiting, or when the median survival is more than 10 years.
Follicular lymphoma in situ and primary follicular lymphoma of the duodenum are particularly indolent variants that rarely progress and rarely require therapy.
A so-called pediatric-type nodal follicular lymphoma has indolent behavior and rarely recurs; adult patients with this histologic variant are characterized by a lack of bcl-2 rearrangement in conjunction with a Ki-67 proliferation index greater than 30% and a localized stage I presentation.
Patients with indolent lymphoma may experience a relapse with a more aggressive histology.
If the clinical pattern of relapse suggests that the disease is behaving in a more aggressive manner, a biopsy should be performed, if feasible.
Documentation of conversion to a more aggressive histology requires an appropriate change to a therapy applicable to that histologic type.
Rapid growth or discordant growth between various disease sites may indicate a histologic conversion.
The risk of histologic transformation was 30% by 10 years in a retrospective review of 325 patients from diagnosis between 1972 and 1999. In this series, high-risk factors for subsequent histologic transformation were advanced stage, high-risk FLIPI, and expectant management.
The 5-year OS rate was more than 50% for patients who had biopsy-proven, aggressive-histology transformation in several multicenter cohort studies employing rituximab plus anthracycline or platinum-based chemotherapy, or similar therapy followed by autologous or allogeneic stem cell transplantation (ASCT).
In a prospective nonrandomized study, at a median follow-up of 6.8 years, 379 (14%) of 2,652 patients subsequently transformed to a more aggressive histology after an initial diagnosis of follicular lymphoma.[Level of evidence: 3iiiDiv] The median OS after subsequent transformation was 5 years; however, among 47 patients with evidence of transformation in conjunction with follicular lymphoma at the time of initial diagnosis, the OS was no worse than that of the other nontransformed patients (5-year OS, 88%; 95% confidence interval [CI], 74%–95%).
Lymphoplasmacytic lymphoma (Waldenström macroglobulinemia)
Lymphoplasmacytic lymphoma is usually associated with a monoclonal serum paraprotein of immunoglobulin M (IgM) type (Waldenström macroglobulinemia).
Most patients have bone marrow, lymph node, and splenic involvement, and some patients may develop hyperviscosity syndrome.
Other lymphomas may also be associated with serum paraproteins.
Asymptomatic patients can be monitored for evidence of disease progression without immediate need for chemotherapy.
Prognostic factors associated with symptoms requiring therapy include the following:
• Age 70 years or older.
• Beta-2-microglobulin of 3 mg/dL or more.
• Increased serum LDH.
Therapeutic approaches
The management of lymphoplasmacytic lymphoma is similar to that of other low-grade lymphomas, especially diffuse, small lymphocytic lymphoma/chronic lymphocytic leukemia.
If the viscosity relative to water is greater than four, the patient may have manifestations of hyperviscosity. Plasmapheresis is useful for temporary, acute symptoms (such as retinopathy, congestive heart failure, and central nervous system [CNS] dysfunction) but should be combined with chemotherapy for prolonged control of the disease.
Symptomatic patients with a serum viscosity of not more than four are usually started directly on chemotherapy.
Therapy may be required to correct hemolytic anemia in patients with chronic cold agglutinin disease; rituximab, cyclophosphamide, and steroids are often employed.
Occasionally, a heated room is required for patients whose cold agglutinins become activated by even minor chilling.
First-line regimens include rituximab, the nucleoside analogs, and alkylating agents, either as single agents or as part of combination chemotherapy.
Rituximab shows 60% to 80% response rates in previously untreated patients, but close monitoring of the serum IgM is required because of a sudden rise in this paraprotein at the start of therapy.[Level of evidence: 3iiiDiv]
The rise of IgM after rituximab can be avoided with the concomitant use of an alkylating agent, such as cyclophosphamide or the proteosome inhibitor bortezomib.
The nucleoside analogs 2-chlorodeoxyadenosine and fludarabine have shown similar response rates for previously untreated patients with lymphoplasmacytic lymphoma.[Level of evidence: 3iiiDiv]
Single-agent alkylators, bendamustine, bortezomib, and combination chemotherapy with or without rituximab also show similar response rates.[Level of evidence: 3iiiDiv]
Currently, no randomized trials guide clinicians about the initial choice of rituximab, nucleoside analogs, alkylating agents, combination chemotherapy, or combinations of these options.
A combination of bortezomib, dexamethasone, and rituximab has been proposed for its high response rate, rapidity of action, and avoidance of an IgM rebound.
In previously treated patients, the B-cell receptor-inhibitor ibrutinib was given to 94 symptomatic patients in two trials, with a response rate of 90% and a PFS of 69% to 86% in 18 to 24 months.[Level of evidence: 3iiiDiv]
Interferon-alpha also shows activity in this disease, in contrast to poor responses in patients with multiple myeloma.
Myeloablative therapy with autologous or allogeneic hematopoietic stem cell support is under clinical evaluation. Candidates for this approach should avoid long-term use of alkylating agents or purine nucleoside analogs, which can deplete hematopoietic stem cells or predispose patients to myelodysplasia or acute leukemia.
After relapse from alkylating-agent therapy, 92 patients with lymphoplasmacytic lymphoma were randomly assigned to either fludarabine or cyclophosphamide, doxorubicin, and prednisone. Although relapse-free survival favored fludarabine (median duration of 19 months vs. 3 months, P < .01), no difference was observed in OS.[Level of evidence: 1iiDii]
Among patients with concomitant hepatitis C virus (HCV) infection, some will attain a complete or partial remission after loss of detectable HCV RNA with treatment using interferon-alpha with or without ribavirin.[Level of evidence: 3iiiDiv]
Marginal zone lymphoma
Marginal zone lymphomas were previously included among the diffuse, small lymphocytic lymphomas.
When marginal zone lymphomas involve the nodes, they are called monocytoid B-cell lymphomas or nodal marginal zone B-cell lymphomas, and when they involve extranodal sites (e.g., gastrointestinal tract, thyroid, lung, breast, orbit, and skin), they are called mucosa-associated lymphatic tissue (MALT) lymphomas.
Gastric MALT
Many patients have a history of autoimmune disease, such as Hashimoto thyroiditis or Sjögren syndrome, or of Helicobacter gastritis.
Most patients present with stage I or stage II extranodal disease, which is most often in the stomach.
Treatment of Helicobacter pylori infection may resolve most cases of localized gastric involvement.
After standard antibiotic regimens, 50% of patients show resolution of gastric MALT by endoscopy after 3 months. Other patients may show resolution after 12 to 18 months of observation.
Of the patients who attain complete remission, 30% demonstrate monoclonality by immunoglobulin heavy chain rearrangement on stomach biopsies with a 5-year median follow-up. The clinical implication of this finding is unknown.
Translocation t(11;18) in patients with gastric MALT predicts for poor response to antibiotic therapy, for H. pylori–negative testing, and for poor response to oral alkylator chemotherapy.
Stable asymptomatic patients with persistently positive biopsies have been successfully followed on a watchful waiting approach until disease progression.
Patients who progress are treated with radiation therapy, rituximab, surgery (total gastrectomy or partial gastrectomy plus radiation therapy), chemotherapy, or combined–modality therapy.
The use of endoscopic ultrasonography may help clinicians to follow responses in these patients. Four case series (encompassing more than 100 patients with stage IE or IIE diffuse, large, B-cell lymphoma with or without associated MALT (but H. pylori-positive) reported durable complete remissions in more than 50% of the patients after treatment of H. pylori.
Extragastric MALT
Localized involvement of other sites can be treated with radiation or surgery.
Patients with extragastric MALT lymphoma have a higher relapse rate than patients with gastric MALT lymphoma in some series, with relapses many years and even decades later. Many of these recurrences involve different MALT sites than the original location.
When disseminated to lymph nodes, bone marrow, or blood, this entity behaves like other low-grade lymphomas.
A prospective, randomized trial of 401 patients with nongastric, extranodal MALT compared chlorambucil alone versus rituximab plus chlorambucil versus rituximab alone. With a median follow-up of 7.4 years, the event-free survival was better for the rituximab-plus-chlorambucil arm (68%) than for the rituximab-alone arm (51%) and for the chlorambucil-alone arm (50%) (P = .0009); however, the 5-year OS was 90% in all arms.
For patients with ocular adnexal MALT, antibiotic therapy using doxycycline that targeted Chlamydia psittaci resulted in durable remissions for almost half of the patients in a review of the literature that included 131 patients.[Level of evidence: 3iiiDiv] These responses to doxycycline are mainly seen in Italian trials and less often in trials conducted in other geographic sites.
Large B-cell lymphomas of MALT sites are classified and treated as diffuse large cell lymphomas.
A large, retrospective review of primary ocular adnexal MALT found that after 10 years of follow-up, 4% of stage I patients treated with radiation therapy transformed to diffuse large B-cell lymphoma, and 3% of them developed CNS involvement.
Nodal marginal zone lymphoma
Patients with nodal marginal zone lymphoma (monocytoid B-cell lymphoma) are treated with the same paradigm of watchful waiting or therapies as described for follicular lymphoma.
Among patients with concomitant HCV infection, the majority attain a complete or partial remission after loss of detectable HCV RNA with treatment using interferon-alpha with or without ribavirin.[Level of evidence: 3iiiDiv]
Mediterranean abdominal lymphoma
The disease variously known as Mediterranean abdominal lymphoma, heavy–chain disease, or immunoproliferative small intestinal disease (IPSID), which occurs in young adults in eastern Mediterranean countries, is another version of MALT lymphoma, which responds to antibiotics in its early stages.
Campylobacter jejuni has been identified as one of the bacterial species associated with IPSID, and antibiotic therapy may result in remission of the disease.
Splenic marginal zone lymphoma
Splenic marginal zone lymphoma is an indolent lymphoma that is marked by massive splenomegaly and peripheral blood and bone marrow involvement, usually without adenopathy.
This type of lymphoma is otherwise known as splenic lymphoma with villous lymphocytes. Splenectomy may result in prolonged remission.
Management is similar to that of other low-grade lymphomas and usually involves rituximab alone or rituximab in combination with purine analogs or alkylating agent chemotherapy.
Splenic marginal zone lymphoma responds less well to chemotherapy, which would ordinarily be effective for chronic lymphocytic leukemia.
Among small numbers of patients with splenic marginal zone lymphoma (splenic lymphoma with villous lymphocytes) and infection with HCV, the majority attained a complete or partial remission after loss of detectable HCV RNA with treatment using interferon-alpha with or without ribavirin.; [Level of evidence: 3iiiDiv] In contrast, no responses to interferon were seen in six HCV-negative patients.
Primary cutaneous anaplastic large cell lymphoma
Primary cutaneous anaplastic large cell lymphoma presents in the skin only with no pre-existing lymphoproliferative disease and no extracutaneous sites of involvement.
Patients with this type of lymphoma encompass a spectrum ranging from clinically benign lymphomatoid papulosis, marked by localized nodules that may regress spontaneously, to a progressive and systemic disease requiring aggressive doxorubicin-based combination chemotherapy.
This spectrum has been called the primary cutaneous CD30-positive T-cell lymphoproliferative disorder.
Patients with localized disease usually undergo radiation therapy.
With more disseminated involvement, watchful waiting or doxorubicin-based combination chemotherapy is applied.
Aggressive NHL
Aggressive NHL includes the following subtypes:
• Diffuse Large B-cell Lymphoma.
• Primary Mediastinal Large B-cell Lymphoma.
• Follicular Large Cell Lymphoma.
• Anaplastic Large Cell Lymphoma.
• Extranodal Natural Killer–/T-cell Lymphoma.
• Lymphomatoid Granulomatosis.
• Angioimmunoblastic T-cell Lymphoma.
• Peripheral T-cell Lymphoma.
• Enteropathy-type Intestinal T-cell Lymphoma.
• Intravascular Large B-cell Lymphoma (Intravascular Lymphomatosis).
• Burkitt Lymphoma/Diffuse Small Noncleaved-cell Lymphoma.
• Lymphoblastic Lymphoma.
• Adult T-cell Leukemia/Lymphoma.
• Mantle Cell Lymphoma.
• Posttransplantation Lymphoproliferative Disorder.
• True Histiocytic Lymphoma.
• Follicular Large Cell Lymphoma.
• Primary Effusion Lymphoma.
• Plasmablastic Lymphoma.
Diffuse Large B-cell Lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most common of the NHL and comprises 30% of newly diagnosed cases.
Most patients present with rapidly enlarging masses, often with both local and systemic symptoms (designated B symptoms with fever, recurrent night sweats, or weight loss).
Some cases of large B-cell lymphoma have a prominent background of reactive T cells and often of histiocytes, so-called T-cell/histiocyte-rich large B-cell lymphoma.
This subtype of large cell lymphoma has frequent liver, spleen, and bone marrow involvement; however, the outcome is equivalent to that of similarly staged patients with DLBCL.
Some patients with DLBCL at diagnosis have a concomitant indolent small B-cell component; while overall survival (OS) appears similar after multidrug chemotherapy, there is a higher risk of indolent relapse.
Prognosis
The vast majority of patients with localized disease are curable with combined–modality therapy or combination chemotherapy alone.
For patients with advanced-stage disease, 50% of presenting patients are cured with doxorubicin-based combination chemotherapy and rituximab.
An International Prognostic Index (IPI) for aggressive NHL (diffuse large cell lymphoma) identifies five significant risk factors prognostic of OS:
1. Age (≤60 years vs. >60 years).
2. Serum lactate dehydrogenase (LDH) (normal vs. elevated).
3. Performance status (0 or 1 vs. 2–4).
4. Stage (stage I or stage II vs. stage III or stage IV).
5. Extranodal site involvement (0 or 1 vs. 2–4).
Patients with two or more risk factors have a less than 50% chance of relapse-free survival and OS at 5 years.
This study also identifies patients at high risk of relapse based on specific sites of involvement, including bone marrow, central nervous system (CNS), liver, lung, and spleen.
Age-adjusted and stage-adjusted modifications of this IPI are used for younger patients with localized disease.
(Møller MB, Christensen BE, Pedersen NT: Prognosis of localized diffuse large B-cell lymphoma in younger patients. Cancer 98 (3): 516-21, 2003. [PUBMED Abstract] http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=12879468&dopt=Abstract)
The BCL-2 gene and rearrangement of the MYC gene or dual overexpression of the MYC gene, or both, confer a particularly poor prognosis. Dose-intensive therapies, infusional therapies, and stem cell transplantation consolidation are being explored in this high-risk group.
A retrospective review evaluated 159 patients with previously untreated DLBCL who underwent double-hit genetic testing by fluorescence in situ hybridization (FISH) and achieved complete response. The induction therapy did not alter 3-year relapse-free survival or OS when autologous stem cell transplantation (ASCT) was employed.
In a retrospective review of 117 patients with relapsed or refractory DLBCL who underwent ASCT, the 4-year OS was 25% for double-hit lymphomas (rearrangement of BCL-2 and MYC), 61% for double-expressor lymphomas (no rearrangement, but increased expression of BCL-2 and MYC), and 70% for patients without these features. Patients at high risk of relapse may be considered for clinical trials.
Molecular profiles of gene expression using DNA microarrays may help to stratify patients in the future for therapies directed at specific targets and to better predict survival after standard chemotherapy.
Patients who have DLBCL with coexpression of CD20 and CD30 may define a subgroup with a unique molecular signature, a more favorable prognosis, and possible therapeutic implication for the use of anti-CD30–specific therapy, such as brentuximab vedotin.
Patients with DLBCL who are event-free after 2 years have a subsequent OS equivalent to that of the age- and sex-matched general population.
CNS prophylaxis
CNS prophylaxis (usually with four to six injections of methotrexate intrathecally) is recommended for patients with testicular involvement.
Some clinicians are employing high-dose intravenous methotrexate (usually four doses) as an alternative to intrathecal therapy because drug delivery is improved and patient morbidity is decreased.
CNS prophylaxis for bone marrow involvement is controversial; some investigators recommend it, others do not.
A retrospective analysis of 605 patients with diffuse large cell lymphoma who did not receive prophylactic intrathecal therapy identified an elevated serum LDH and more than one extranodal site as independent risk factors for CNS recurrence.
Patients with both risk factors have a 17% probability of CNS recurrence at 1 year after diagnosis (95% confidence interval [CI], 7%–28%) versus 2.8% (95% CI, 2.7%–2.9%) for the remaining patients.[Level of evidence: 3iiiDiii]
The CNS-International Prognostic Index (IPI) is a tool used to predict which patients have a risk of CNS relapse above 10%. It was developed by the German Lymphoma Study Group and validated by the British Columbia Cancer Agency database.
(Schmitz N, Zeynalova S, Nickelsen M, et al.: CNS International Prognostic Index: A Risk Model for CNS Relapse in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP. J Clin Oncol 34 (26): 3150-6, 2016. [PUBMED Abstract] http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=27382100&dopt=Abstract)
Four to six of the IPI risk factors (refer to the Prognosis section for more information) and the involvement of the kidneys or adrenal glands were used to define the high-risk group that might benefit from CNS prophylaxis.
The addition of rituximab to cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP)-based regimens has significantly reduced the risk of CNS relapse in retrospective analyses. Patients with CNS dissemination at diagnosis or at relapse usually receive rituximab and high-doses of methotrexate and/or cytarabine followed by ASCT, but this approach has not been assessed in randomized trials.[Level of evidence: 3iiiDiv]
Primary Mediastinal Large B-cell Lymphoma
Primary mediastinal (thymic) large B-cell lymphoma is a subset of DLBCL with molecular characteristics that are most similar to nodular-sclerosing Hodgkin lymphoma (HL).
Mediastinal lymphomas with features intermediate between primary mediastinal B-cell lymphoma and nodular-sclerosing HL are called mediastinal gray-zone lymphomas.
Patients are usually female and young (median age, 30–40 years).
Patients present with a locally invasive anterior mediastinal mass that may cause respiratory symptoms or superior vena cava syndrome.
Prognosis and therapy is the same as for other comparably staged patients with DLBCL.
Uncontrolled, phase II studies employing dose-adjusted EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) plus rituximab or R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) show high cure rates while avoiding any mediastinal radiation.[Level of evidence: 3iiiA]
These results suggest that patients who receive R-CHOP-based regimens may avoid the serious long-term complications of radiation therapy when given with chemotherapy.
Posttreatment fluorine F 18-fludeoxyglucose (18F) positron emission tomography–computed tomography (18F-FDG PET-CT) scans appear unreliable with many false positives.
According to an anecdotal prospective single-arm trial, describing a posttherapy-positive 18F-FDG PET-CT as greater than liver uptake (rather than mediastinal blood-pool uptake) may identify patients with an increased risk of relapse.
The only randomized trial showing an OS advantage for combined modality therapy was retracted.
Follicular Large Cell Lymphoma
Prognosis
The natural history of follicular large cell lymphoma remains controversial. While there is agreement about the significant number of long-term disease-free survivors with early-stage disease, the curability of patients with advanced disease (stage III or stage IV) remains uncertain.
Some groups report a continuous relapse rate similar to the other follicular lymphomas (a pattern of indolent lymphoma). Other investigators report a plateau in freedom from progression at levels expected for an aggressive lymphoma (40% at 10 years).
This discrepancy may be caused by variations in histologic classification between institutions and the rarity of patients with follicular large cell lymphoma.
A retrospective review of 252 patients, all treated with anthracycline-containing combination chemotherapy, showed that patients with more than 50% diffuse components on biopsy had a worse OS than other patients with follicular large cell lymphoma.
Therapeutic approaches
Treatment of follicular large cell lymphoma is more similar to treatment of aggressive NHL than it is to the treatment of indolent NHL.
In support of this approach, treatment with high-dose chemotherapy and autologous hematopoietic peripheral stem cell transplantation (SCT) shows the same curative potential in patients with follicular large cell lymphoma who relapse as it does in patients with diffuse large cell lymphoma who relapse.[Level of evidence: 3iiiA]
Anaplastic Large Cell Lymphoma
Anaplastic large cell lymphomas (ALCL) may be confused with carcinomas and are associated with the Ki-1 (CD30) antigen.
These lymphomas are usually of T-cell origin, often present with extranodal disease, and are found especially in the skin.
The translocation of chromosomes 2 and 5 creates a unique fusion protein with a nucleophosmin-anaplastic lymphoma kinase (ALK).
Patients whose lymphomas express ALK (immunohistochemistry) are usually younger and may have systemic symptoms, extranodal disease, and advanced-stage disease; however, they have a more favorable survival rate than that of ALK-negative patients.
Patients with ALK-positive ALCL are generally treated the same as patients with diffuse large cell lymphomas using the CHOP regimen and have a prognosis that is as good as that for comparably staged patients.
For patients with relapsed disease, anecdotal responses have been reported for brentuximab vedotin (anti-tubulin agent attached to a CD30-specific monoclonal antibody), romidepsin, and pralatrexate.[Level of evidence: 3iiiDiv]
For patients with relapsed disease, ASCT showed a 50% 3-year progression-free survival (PFS) for 39 patients in a retrospective review.[Level of evidence: 3iiiDiii]
ALCL in children is usually characterized by systemic and cutaneous disease and has high response rates and good OS with doxorubicin-based combination chemotherapy.
Patients with breast implant–associated ALCL may do well without chemotherapy after capsulectomy and implant removal if the disease is confined to the fibrous capsule, and no associated mass is present.
Extranodal Natural Killer–/T-cell Lymphoma
Extranodal natural killer (NK)–/T-cell lymphoma (nasal type) is an aggressive lymphoma marked by extensive necrosis and angioinvasion, most often presenting in extranodal sites, in particular the nasal or paranasal sinus region.
Other extranodal sites include the palate, trachea, skin, and gastrointestinal tract.
Hemophagocytic syndrome may occur; historically, these tumors were considered part of lethal midline granuloma.
In most cases, Epstein-Barr virus (EBV) genomes are detectable in the tumor cells and immunophenotyping shows CD56 positivity.
Cases with blood and marrow involvement are considered NK-cell leukemia.
The increased risk of CNS involvement and of local recurrence has led to recommendations for radiation therapy locally, concurrently, or before the start of chemotherapy, and for intrathecal prophylaxis and/or prophylactic cranial radiation therapy.
A retrospective review of 1,273 early-stage patients stratified patients into a low-risk group and high-risk group using stage, age, LDH, performance status, and primary tumor invasion. Low-risk patients fared best with radiation therapy alone, while high-risk patients fared best with a strategy of radiation therapy followed by chemotherapy.
The highly aggressive course, with poor response and short survival with standard therapies, especially for patients with advanced-stage disease or extranasal presentation, has led some investigators to recommend autologous or allogeneic peripheral SCT consolidation.
L-asparaginase-containing regimens have shown anecdotal response rates greater than 50% for relapsing, refractory, or newly diagnosed stage IV patients.
NK-/T-cell lymphoma that presents only in the skin has a more favorable prognosis, especially in patients with coexpression of CD30 with CD56.
A benign NK-cell enteropathy (EBV negative) on endoscopic biopsy should be distinguished from NK-/T-cell lymphoma.
Lymphomatoid Granulomatosis
Lymphomatoid granulomatosis is an EBV-positive large B-cell lymphoma with a predominant T-cell background.
The histology shows association with angioinvasion and vasculitis, usually manifesting as pulmonary lesions or paranasal sinus involvement.
Patients are managed like others with diffuse large cell lymphoma and require doxorubicin-based combination chemotherapy.
Angioimmunoblastic T-cell Lymphoma
Angioimmunoblastic T-cell lymphoma (AITL) or (ATCL) was formerly called angioimmunoblastic lymphadenopathy with dysproteinemia.
Characterized by clonal T-cell receptor gene rearrangement, this entity is managed like diffuse large cell lymphoma.
Patients present with profound lymphadenopathy, fever, night sweats, weight loss, skin rash, a positive Coombs test, and polyclonal hypergammaglobulinemia.
Opportunistic infections are frequent because of an underlying immune deficiency.
B-cell EBV genomes are detected in most affected patients.
Doxorubicin-based combination chemotherapy, such as the CHOP regimen, is recommended as it is for other aggressive lymphomas.
The International Peripheral T-Cell Lymphoma Project involving 22 international centers identified 243 patients with AITL or ATCL; the 5-year OS and failure-free survival rates were 33% and 18%, respectively.
Myeloablative chemotherapy and radiation therapy with autologous or allogeneic peripheral stem cell support has been described in anecdotal reports.
Anecdotal responses have been reported for cyclosporine, pralatrexate, bendamustine, the histone deacetylase inhibitor romidepsin, and brentuximab vedotin (even if there is little or no CD30 expression on the lymphoma).[Level of evidence: 3iiiDiv]
Occasional spontaneous remissions and protracted responses to steroids only have been reported.
Peripheral T-cell Lymphoma
Patients with peripheral T-cell lymphoma have diffuse large cell or diffuse mixed lymphoma that expresses a cell surface phenotype of a postthymic (or peripheral) T-cell expressing CD4 or CD8 but not both together.
Peripheral T-cell lymphoma encompasses a group of heterogeneous nodal T-cell lymphomas that will require future delineation. This includes the so-called Lennert lymphoma, a T-cell lymphoma admixed with a preponderance of lymphoepithelioid cells.
Prognosis
Most investigators report worse response and survival rates for patients with peripheral T-cell lymphomas than for patients with comparably staged B-cell aggressive lymphomas.
Most patients present with multiple adverse prognostic factors (i.e., older age, stage IV, multiple extranodal sites, and elevated LDH), and these patients have a low (<20%) failure-free survival and OS at 5 years.
Therapeutic approaches
Therapy involves doxorubicin-based combination chemotherapy (such as CHOP or CHOPE [CHOP plus etoposide]), which is also used for DLBCL.
For patients with early-stage disease, anecdotal retrospective series disagree on the value of consolidative radiation therapy after combination chemotherapy.[Level of evidence: 3iiiDiv]
Consolidation using high-dose chemotherapy with autologous or allogeneic hematopoietic stem cell support has been applied to patients with advanced-stage peripheral T-cell lymphoma after induction therapy with CHOP-based regimens and after response to reinduction therapy at first relapse. Evidence for this approach is anecdotal.
For relapsing patients, pralatrexate has shown a 30% response rate and a median 10-month duration of response for 109 evaluable patients in a prospective trial.[Level of evidence: 3iiiDiv]
Also for relapsing patients, similar response rates were seen for romidepsin for 130 evaluable patients in a prospective trial.[Level of evidence: 3iiiDiv]
Anecdotal responses have been seen with pralatrexate, bendamustine, belinostat, and brentuximab vedotin (even if there is little or no CD30 expression on the lymphoma).[Level of evidence: 3iiiDiv]
Incorporation of these new agents with CHOP chemotherapy is under clinical evaluation.
Anecdotal responses have also been seen with alemtuzumab, an anti-CD52 monoclonal antibody, after relapse from previous chemotherapy.
The median PFS after first relapse was less than 6 months in one series of 163 patients with peripheral T-cell lymphoma.
An unusual type of peripheral T-cell lymphoma occurring mostly in young men, hepatosplenic T-cell lymphoma, appears to be localized to the hepatic and splenic sinusoids, with cell surface expression of the T-cell receptor gamma/delta.
Another variant, subcutaneous panniculitis-like T-cell lymphoma, is localized to subcutaneous tissue associated with hemophagocytic syndrome. These patients have cells that express alpha-beta phenotype.
Those with gamma-delta phenotype have a more aggressive clinical course and are classified as cutaneous gamma-delta T-cell lymphoma.
These patients may manifest involvement of the epidermis, dermis, subcutaneous region, or mucosa. These entities have extremely poor prognoses with an extremely aggressive clinical course and are treated within the same paradigm as the highest-risk groups with DLBCL.
An indolent T-cell lymphoproliferative disease of the gastrointestinal tract must be distinguished from peripheral T-cell lymphoma because no therapy may be indicated.
Enteropathy-type Intestinal T-cell Lymphoma
Enteropathy-type intestinal T-cell lymphoma involves the small bowel of patients with gluten-sensitive enteropathy (celiac sprue).
Because a gluten-free diet prevents the development of lymphoma, patients diagnosed with celiac sprue in childhood rarely develop lymphoma.
The diagnosis of celiac disease is usually made by finding villous atrophy in the resected intestine.
Surgery is often required for diagnosis and to avoid perforation during therapy.
Therapy is with doxorubicin-based combination chemotherapy, but relapse rates appear higher than for comparably staged diffuse large cell lymphoma.
Complications of treatment include gastrointestinal bleeding, small bowel perforation, and enterocolic fistulae; patients often require parenteral nutrition.
Multifocal intestinal perforations and visceral abdominal involvement are seen at the time of relapse.
High-dose therapy with hematopoietic stem cell rescue has been applied in first remission or at relapse.[Level of evidence: 3iiiDiii] Evidence for this approach is anecdotal.
Intravascular Large B-cell Lymphoma (Intravascular Lymphomatosis)
Intravascular lymphomatosis is characterized by large cell lymphoma confined to the intravascular lumen.
The brain, kidneys, lungs, and skin are the organs most likely affected by intravascular lymphomatosis.
With the use of aggressive combination chemotherapy, the prognosis is similar to more conventional presentations.
Burkitt Lymphoma/Diffuse Small Noncleaved-cell Lymphoma
Burkitt lymphoma/diffuse small noncleaved-cell lymphoma typically involves younger patients and represents the most common type of pediatric NHL.
These types of aggressive extranodal B-cell lymphomas are characterized by translocation and deregulation of the C-Myc gene on chromosome 8.
A subgroup of patients with dual translocation of C-Myc and bcl-2 appear to have an extremely poor outcome despite aggressive therapy (5-month OS).[Level of evidence: 3iiiA]
In some patients with larger B cells, there is morphologic overlap with DLBCL.
These Burkitt-like large cell lymphomas show C-Myc deregulation, extremely high proliferation rates, and a gene-expression profile as expected for classic Burkitt lymphoma.
Endemic cases, usually from Africa, involve the facial bones or jaws of children, mostly containing EBV genomes.
Sporadic cases usually involve the gastrointestinal system, ovaries, or kidneys.
Patients present with rapidly growing masses and a very high LDH but are potentially curable with intensive doxorubicin-based combination chemotherapy.
Therapeutic approaches
Treatment of Burkitt lymphoma/diffuse small noncleaved-cell lymphoma involves aggressive multidrug regimens in combination with rituximab, similar to those used for the advanced-stage aggressive lymphomas (diffuse large cell).
Aggressive combination chemotherapy, which is patterned after that used in childhood Burkitt lymphoma, has been very successful for adult patients with more than 60% of advanced-stage patients free of disease at 5 years.
Adverse prognostic factors include bulky abdominal disease and high serum LDH.
Patients with Burkitt lymphoma have a 20% to 30% lifetime risk of CNS involvement.
Prophylaxis with intrathecal chemotherapy is required as part of induction therapy.
Patients with HIV-associated Burkitt lymphoma also benefit from less-toxic modification of the aggressive multidrug regimens in combination with rituximab.[Level of evidence: 3iiiDiv]
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (precursor T-cell) is a very aggressive form of NHL.
It often, but not exclusively, occurs in young patients.
It is commonly associated with large mediastinal masses and has a high predilection for disseminating to bone marrow and the CNS.
Careful review of the pathologic specimens, bone marrow aspirate, biopsy specimen, cerebrospinal fluid cytology, and lymphocyte marker constitute the most important aspects of the pretreatment staging workup.
Treatment is usually patterned after that for acute lymphoblastic leukemia.
Intensive combination chemotherapy with or without bone marrow transplantation is the standard treatment for this aggressive histologic type of NHL.
Radiation therapy is sometimes given to areas of bulky tumor masses.
Because these forms of NHL tend to progress quickly, combination chemotherapy is instituted rapidly once the diagnosis has been confirmed.
Adult T-cell Leukemia/Lymphoma
Adult T-cell leukemia/lymphoma (ATL) is caused by infection with the retrovirus human T-lymphotrophic virus 1 and is frequently associated with lymphadenopathy, hypercalcemia, circulating leukemic cells, bone and skin involvement, hepatosplenomegaly, a rapidly progressive course, and poor response to combination chemotherapy.
ATL has been divided into four clinical subtypes:
• Acute (aggressive course with leukemia with or without extranodal or nodal involvement).
• Lymphoma (aggressive course with lymphadenopathy and no leukemia).
• Chronic (indolent course with leukemia and lymphadenopathy).
• Smoldering (indolent course with only leukemia).
The acute and lymphoma types of ATL have done poorly with strategies of combination chemotherapy and allogeneic SCT (alloSCT) with a median OS under 1 year.
Using combination chemotherapy, less than 10% of 807 patients were alive after 4 years.
Anecdotal durable remissions have been reported after alloSCT and even after subsequent donor lymphocyte infusion for relapses after transplant.[Level of evidence: 3iiiDiv]
Among 815 patients who underwent alloSCT in two retrospective reviews, the 3-year OS rates were 36% and 26%.[Level of evidence: 3iiiA]
The combination of zidovudine and interferon-alpha has activity against ATL, even for patients who failed previous cytotoxic therapy.
Durable remissions are seen in the majority of presenting patients with this combination but are not seen in patients with the lymphoma subtype of ATL.
In a multicenter phase II study of 26 relapsed patients, 42% responded to lenalidomide (including four complete responses).[Level of evidence: 3iiiDiv]
Symptomatic local progression of all subtypes responds well to palliative radiation therapy.
Mantle Cell Lymphoma
Mantle cell lymphoma is found in lymph nodes, the spleen, bone marrow, blood, and sometimes the gastrointestinal system (lymphomatous polyposis).
Mantle cell lymphoma is characterized by CD5-positive follicular mantle B cells, a translocation of chromosomes 11 and 14, and an overexpression of the cyclin D1 protein.
Like the low-grade lymphomas, mantle cell lymphoma appears incurable with anthracycline-based chemotherapy and occurs in older patients with generally asymptomatic advanced-stage disease.
The median survival, however, is significantly shorter (5–7 years) than that of other lymphomas, and this histology is now considered to be an aggressive lymphoma.
A diffuse pattern and the blastoid variant have an aggressive course with shorter survival, while the mantle zone type may have a more indolent course.
A high cell-proliferation rate (increased Ki-67, mitotic index, beta-2-microglobulin) may be associated with a poorer prognosis.
Therapeutic approaches
Asymptomatic patients with low-risk scores on the IPI may do well when initial therapy is deferred.[Level of evidence: 3iiiDiv]
There is no standard approach to mantle cell lymphoma. Several induction chemotherapy regimens may be employed for symptomatic progressing disease.
These regimens range in intensity from rituximab alone to rituximab plus bendamustine, to R-CHOP, to high-dose intensive regimens such as R-hyper C-VAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with methotrexate and cytarabine).
Some physicians use autologous or ASCT consolidation next, while others prefer rituximab maintenance, reserving high-dose consolidation for a later time.
Ibrutinib, lenalidomide, and bortezomib have shown activity in relapsing patients, and these drugs are being incorporated upfront.
It is unclear which therapeutic approach offers the best long-term survival in this clinicopathologic entity.
In a prospective randomized trial, 532 patients older than 60 years and not eligible for SCT were given either R-CHOP or R-FC (rituximab, fludarabine, cyclophosphamide) for 6 to 8 cycles, followed by maintenance therapy in responders randomly assigned to rituximab or interferon-alpha maintenance therapy.
With a median follow-up of 37 months, the OS was significantly shorter after R-FC than after R-CHOP (47% vs. 62%, P = .005; hazard ratio [HR]death, 1.50; 95% CI, 1.13–1.99).[Level of evidence: 1iiA]
Event-free survival favored rituximab over interferon-alpha (57% PFS at 4 years vs. 34%, P = .01; HR, 0.55; 95% CI, 0.36–0.87), but OS did not differ significantly (79% vs. 67% at 4 years, P = .13).[Level of evidence: 1iiDi]
However, patients who received R-CHOP induction showed an OS benefit for rituximab maintenance over interferon-alpha maintenance (87% vs. 63% at 4 years, P = .005).[Level of evidence: 3iiiA]
A randomized trial compared bendamustine plus rituximab with R-CHOP and showed improved PFS (35 vs. 22 months; HR, 0.49; 95% CI, 0.28–0.79; P = .004) but no difference in OS.[Level of evidence: 1iiDiii]
A prospective, randomized trial of 487 patients compared VR-CAP (bortezomib, rituximab, cyclophosphamide, doxorubicin, prednisone) with R-CHOP. With a median follow-up of 40 months, the median PFS favored VR-CAP (24.7 months vs. 14.4 months, HR, 0.63; P <.001), but the 4-year OS was not significantly different (64% vs. 54%, P = .17).[Level of evidence: 1iiDiii]
A prospective randomized trial of 497 patients younger than 65 years compared six cycles of R-CHOP to six cycles of alternating R-CHOP and R-DHAP (rituximab, dexamethasone, cytarabine, and cisplatin), with both groups then receiving autologous stem cell transplantation.[Level of evidence: 1iiDiii] With a median follow-up of 6.1 years, the time to treatment failure (TTF) was longer in the cytarabine group, with a median TTF follow-up of 9.1 years (95% CI, 6.3 years to not reached) compared with 3.9 years (95% CI, 3.2‒4.4 years) (HR, 0.56; corrected P = .038) for the control group. Despite this surprising difference in TTF, the OS was not different.
Many investigators are exploring high-dose chemoradiation immunotherapy with stem cell/marrow support or nonmyeloablative ASCT.
Thus far, randomized trials have not shown OS benefits from these newer approaches.
Lenalidomide with or without rituximab also shows response rates of around 50% in relapsed patients, with even higher response rates for previously untreated patients.[Level of evidence: 3iiDiv]
The B-cell receptor-inhibitor ibrutinib showed a response rate of 86% (21% complete response) in previously treated patients with a median PFS of 14 months.[Level of evidence: 3iiiDiv]
Posttransplantation Lymphoproliferative Disorder
Patients who undergo transplantation of the heart, lung, liver, kidney, or pancreas usually require lifelong immunosuppression.
This may result in posttransplantation lymphoproliferative disorder (PTLD) in 1% to 3% of recipients, which appears as an aggressive lymphoma.
Pathologists can distinguish a polyclonal B-cell hyperplasia from a monoclonal B-cell lymphoma; both are almost always associated with EBV.
Prognosis
Poor performance status, grafted organ involvement, high IPI, elevated LDH, and multiple sites of disease are poor prognostic factors for PTLD.
Therapeutic options
In some cases, withdrawal of immunosuppression results in eradication of the lymphoma.
When this is unsuccessful or not feasible, a trial of rituximab may be considered, because it has shown durable remissions in approximately 60% of patients and a favorable toxicity profile.
If these measures fail, doxorubicin-based combination chemotherapy (R-CHOP) is recommended, although some patients can avoid cytotoxic therapy.
Localized presentations can be controlled with surgery or radiation therapy alone. These localized mass lesions, which may grow over a period of months, are often phenotypically polyclonal and tend to occur within weeks or a few months after transplantation.
Multifocal, rapidly progressive disease occurs late after transplantation (>1 year) and is usually phenotypically monoclonal and associated with EBV.
These patients may have durable remissions using standard chemotherapy regimens for aggressive lymphoma.
Instances of EBV-negative PTLD occur even later (median, 5 years posttransplant) and have a worse prognosis; R-CHOP chemotherapy should be applied directly in this circumstance.
A sustained clinical response after failure from chemotherapy was attained using an immunotoxin (anti-CD22 B-cell surface antigen antibody linked with ricin, a plant toxin).
An anti-interleukin-6 monoclonal antibody is also under clinical evaluation.
True Histiocytic Lymphoma
True histiocytic lymphomas are very rare tumors that show histiocytic differentiation and express histiocytic markers in the absence of B-cell or T-cell lineage-specific immunologic markers.
Care must be taken with immunophenotypic tests to exclude ALCL or hemophagocytic syndromes caused by viral infections, especially EBV.
Therapeutic options
Therapy is modeled after the treatment of comparably staged diffuse large cell lymphomas, but the optimal approach remains to be defined.
Primary Effusion Lymphoma
Primary effusion lymphoma presents exclusively or mainly in the pleural, pericardial, or abdominal cavities in the absence of an identifiable tumor mass.
Patients are usually human immunodeficiency virus seropositive, and the tumor usually contains Kaposi sarcoma–associated herpes virus/human herpes virus 8.
Prognosis
The prognosis of primary effusion lymphoma is extremely poor.
Therapeutic approaches
Therapy is usually modeled after the treatment of comparably staged diffuse large cell lymphomas.
Plasmablastic Lymphoma
Plasmablastic lymphoma is most often seen in patients with HIV infection and is characterized by CD20-negative large B cells with plasmacytic features.
This type of lymphoma has a very aggressive clinical course, including poor responses and short remissions with standard chemotherapy.
Anecdotal reports suggest using aggressive chemotherapy for Burkitt or lymphoblastic lymphoma, followed by stem cell transplant consolidation in responding patients, when feasible.
Stage Information for Adult NHL
Stage is important in selecting a treatment for patients with non-Hodgkin lymphoma (NHL).
Chest and abdominal computed tomography (CT) scans are usually part of the staging evaluation for all lymphoma patients.
The staging system is similar to the staging system used for Hodgkin lymphoma (HL).
Common among patients with NHL is involvement of the following:
• Noncontiguous lymph nodes.
• Waldeyer ring.
• Epitrochlear nodes.
• Gastrointestinal tract.
• Extranodal presentations. (A single extranodal site is occasionally the only site of involvement in patients with diffuse lymphoma.)
• Bone marrow.
• Liver (especially common in patients with low-grade lymphomas).
Cytologic examination of cerebrospinal fluid may be positive in patients with aggressive NHL.
Involvement of hilar and mediastinal lymph nodes is less common than in HL.
Mediastinal adenopathy, however, is a prominent feature of lymphoblastic lymphoma and primary mediastinal B-cell lymphoma, entities primarily found in young adults.
The majority of patients with NHL present with advanced (stage III or stage IV) disease that can often be identified with limited staging procedures such as CT scanning and biopsies of the bone marrow and other accessible sites of involvement.
Laparoscopic biopsy or laparotomy is not required for staging but may be necessary to establish a diagnosis or histologic type.
Positron emission tomography (PET) with fluorine F 18-fludeoxyglucose can be used for initial staging and for follow-up after therapy as a supplement to CT scanning.
Interim PET scans after two to four cycles of therapy did not provide reliable prognostic information because of problems of interobserver reproducibility in a large cooperative group trial (ECOG-E344 [NCT00274924]) and lack of difference in outcome between PET-negative and PET-positive/biopsy-negative patients in two prospective trials and in a meta-analysis.
For patients with follicular lymphoma, a positive PET result after therapy has a worse prognosis; however, it is unclear whether a positive PET result is predictive when further or different therapy is implemented.
In a retrospective study of 130 patients with diffuse large B-cell lymphoma, PET scanning identified all clinically important marrow involvement from lymphoma, and bone marrow biopsy did not upstage any patient.
Bone marrow biopsies are required for some clinical trials and when the identification of marrow involvement would change the therapeutic plan.
Staging Subclassification System
| Stage | Prognostic Groups |
| I |
Involvement of a single lymphatic site (i.e., nodal region, Waldeyer ring, thymus or spleen) (I). OR Localized involvement of a single extralymphatic organ or site in the absence of any lymph node involvement (IE) (rare in Hodgkin lymphoma). |
| II |
Involvement of two or more lymph node regions on the same side of the diaphragm (II). OR Localized involvement of a single extralymphatic organ or site in association with regional lymph node involvement with or without involvement of other lymph node regions on the same side of the diaphragm (IIE). The number of regions involved may be indicated by a subscript Arabic numeral, for example, II3. |
| III | Involvement of lymph node regions on both sides of the diaphragm (III), which also may be accompanied by extralymphatic extension in association with adjacent lymph node involvement (IIIE) or by involvement of the spleen (IIIS) or both (IIIE, IIIS). Splenic involvement is designated by the letter S. |
| IV |
Diffuse or disseminated involvement of one or more extralymphatic organs, with or without associated lymph node involvement. OR Isolated extralymphatic organ involvement in the absence of adjacent regional lymph node involvement, but in conjunction with disease in distant site(s). Stage IV includes any involvement of the liver or bone marrow, lungs (other than by direct extension from another site), or cerebrospinal fluid. |
The Ann Arbor staging system is commonly used for patients with NHL. In this system, stage I, stage II, stage III, and stage IV adult NHL can be subclassified into A and B categories: B for those with well-defined generalized symptoms and A for those without such symptoms.
The B designation is given to patients with any of the following symptoms:
• Unexplained loss of more than 10% of body weight in the 6 months before diagnosis.
• Unexplained fever with temperatures above 38°C.
• Drenching night sweats.
The Lugano classification system eliminates the A and B categories for staging of NHL.
Occasionally, specialized staging systems are used. The physician should be aware of the system used in a specific report.
The E designation is used when extranodal lymphoid malignancies arise in tissues separate from, but near, the major lymphatic aggregates.
Stage IV refers to disease that is diffusely spread throughout an extranodal site, such as the liver. If pathologic proof of involvement of one or more extralymphatic sites has been documented, the symbol for the site of involvement, followed by a plus sign (+), is listed.
Notation to Identify Specific Sites
N = nodes
H = liver
L = lung
M = bone marrow
S = spleen
P = pleura
O = bone
D = skin
Current practice assigns a clinical stage based on the findings of the clinical evaluation and a pathologic stage based on the findings made as a result of invasive procedures beyond the initial biopsy.
For example, on percutaneous biopsy, a patient with inguinal adenopathy and a positive lymphangiogram without systemic symptoms might be found to have involvement of the liver and bone marrow. The precise stage of such a patient would be clinical stage IIA, pathologic stage IVA(H+)(M+).
A number of other factors that are not included in the above staging system are important for the staging and prognosis of patients with NHL.
These factors include the following:
• Age.
• Performance status (PS).
• Tumor size.
• Lactate dehydrogenase (LDH) values.
• The number of extranodal sites.
To identify subgroups of patients most likely to relapse, an international prognostic index was compiled for 2,031 patients with aggressive NHL. After validation by several cancer centers, the major cooperative groups have used this index in the design of new clinical trials. The model is simple to apply, reproducible, and predicts outcome even after patients have achieved a complete remission. The model identifies five significant risk factors prognostic of overall survival (OS):
• Age (<60 years vs. >60 years).
• Serum LDH (normal vs. elevated).
• PS (0 or 1 vs. 2–4).
• Stage (stage I or stage II vs. stage III or stage IV).
• Extranodal site involvement (0 or 1 vs. 2–4).
Patients with two or more risk factors have a less than 50% chance of relapse-free survival and OS at 5 years.
This study also identifies patients at high risk of relapse based on specific sites of involvement, including bone marrow, central nervous system, liver, lung, and spleen.
The bcl-2 gene and rearrangement of the myc gene or dual overexpression of the myc gene, or both, confer a particularly poor prognosis.
Patients at high risk of relapse may benefit from consolidation therapy or other approaches under clinical evaluation.
Molecular profiles of gene expression using DNA microarrays may help to stratify patients in the future for therapies directed at specific targets and to better predict survival after standard chemotherapy.
Treatment Option Overview for Adult NHL
Treatment of non-Hodgkin lymphoma (NHL) depends on the histologic type and stage.
Many of the improvements in survival have been made using clinical trials (experimental therapy) that have attempted to improve on the best available accepted therapy (conventional or standard therapy).
In asymptomatic patients with indolent forms of advanced NHL, treatment may be deferred until the patient becomes symptomatic as the disease progresses.
When treatment is deferred, the clinical course of patients with indolent NHL varies; frequent and careful observation is required so that effective treatment can be initiated when the clinical course of the disease accelerates.
Some patients have a prolonged indolent course, but others have disease that rapidly evolves into more aggressive types of NHL that require immediate treatment.
Radiation techniques differ somewhat from those used in the treatment of Hodgkin lymphoma.
The dose of radiation therapy usually varies from 25 Gy to 50 Gy and is dependent on factors that include the histologic type of lymphoma, the patient’s stage and overall condition, the goal of treatment (curative or palliative), the proximity of sensitive surrounding organs, and whether the patient is being treated with radiation therapy alone or in combination with chemotherapy.
Given the patterns of disease presentations and relapse, treatment may need to include unusual sites such as Waldeyer ring, epitrochlear, or mesenteric nodes. The associated morbidity of the treatment must be considered carefully.
The majority of patients who receive radiation are usually treated on only one side of the diaphragm.
Localized presentations of extranodal NHL may be treated with involved-field techniques with significant (>50%) success.
| Stage | Standard Treatment Options |
| Indolent, Stage I and Contiguous Stage II Adult NHL | Radiation therapy |
| Rituximab with or without chemotherapy | |
| Watchful waiting | |
| Other therapies as designated for patients with advanced-stage disease | |
| Indolent, Noncontiguous Stage II/III/IV Adult NHL | Watchful waiting for asymptomatic patients |
| Rituximab | |
| Idelalisib | |
| Purine nucleoside analogs | |
| Alkylating agents (with or without steroids) | |
| Combination chemotherapy | |
| Yttrium Y 90-ibritumomab tiuxetan | |
| Maintenance rituximab | |
| Indolent, Recurrent Adult NHL | Chemotherapy (single agent or combination) |
| Rituximab | |
| Lenalidomide | |
| Radiolabeled anti-CD20 monoclonal antibodies | |
| Palliative radiation therapy | |
| Aggressive, Stage I and Contiguous Stage II Adult NHL | R-CHOP with or without IF-XRT |
| Aggressive, Noncontiguous Stage II/III/IV Adult NHL | R-CHOP |
| Other combination chemotherapy | |
| Adult Lymphoblastic Lymphoma | Intensive therapy |
| Radiation therapy | |
| Diffuse, Small, Noncleaved-Cell/Burkitt Lymphoma | Aggressive multidrug regimens |
| Central nervous system (CNS) prophylaxis | |
| Aggressive, Recurrent Adult NHL | Bone marrow or stem cell transplantation |
| Re-treatment with standard agents | |
| Palliative radiation therapy |
CHOP = cyclophosphamide, doxorubicin, vincristine, prednisone;
R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone;
IF-XRT = involved-field radiation therapy;
Even though standard treatment in patients with lymphomas can cure a significant fraction, numerous clinical trials that explore improvements in treatment are in progress.
If possible, patients should be included in these studies. Standardized guidelines for response assessment have been suggested for use in clinical trials.
Several retrospective reviews suggest routine surveillance scans after attaining clinical complete remission after induction therapy for diffuse large B-cell lymphoma offer little to no value.
Prognostic value is also difficult to identify for an interim positron emission tomography-computed tomography scan during induction therapy for diffuse large B-cell lymphoma.
Aggressive lymphomas are increasingly seen in human immunodeficiency virus (HIV)-positive patients; treatment of these patients requires special consideration.
In addition to screening for HIV among patients with aggressive lymphomas, active hepatitis B or hepatitis C should be assessed before treatment with rituximab and/or chemotherapy. Even patients with undetectable hepatitis B viral loads after remote past infection benefit from prophylaxis with entecavir in the context of rituximab therapy.
Similarly, prophylaxis for herpes zoster with acyclovir or valacyclovir and prophylaxis for pneumocystis with trimethoprim/sulfamethoxazole or dapsone are usually applied with rituximab with or without combination chemotherapy.
Several unusual presentations of lymphoma occur that often require somewhat modified approaches to staging and therapy. The reader is referred to reviews for a more detailed description of extranodal presentations in the gastrointestinal system, thyroid, spleen, testis, paranasal sinuses, bone, orbit, and skin.
Treatment for Indolent, Stage I and Contiguous Stage II Adult NHL
Although localized presentations are uncommon in non-Hodgkin lymphoma (NHL), the goal of treatment should be to cure the disease in patients who are shown to have truly localized occurrence after undergoing appropriate staging procedures.
Standard Treatment Options for Indolent, Stage I and Contiguous Stage II Adult NHL
1. Radiation therapy.
2. Rituximab with or without chemotherapy.
3. Watchful waiting.
4. Other therapies as designated for patients with advanced-stage disease.
The National Lymphocare Study identified 471 patients with stage I follicular lymphoma.
Of those patients, 206 were rigorously staged with a bone marrow aspirate and biopsy, and computed tomography (CT) scans or positron emission tomography (PET-CT) scans.
Nonrandomized treatments included radiation therapy (27%), rituximab-chemotherapy (R-chemotherapy) (28%), watchful waiting (17%), R-chemotherapy plus radiation therapy (13%), and rituximab alone (12%), although more than one-third of the patients started with expectant therapy.
With a median follow-up of 57 months, progression-free survival favored R-chemotherapy or R-chemotherapy plus radiation therapy, but overall survival was nearly identical, all over 90%.[Level of evidence: 3iiiD]
Clinical trials are required to answer questions such as:
• If the PET-CT scan is clear after excisional biopsy, is watchful waiting or radiation therapy preferred?
• Should rituximab be added to radiation therapy for stage I follicular lymphoma?
• Is there any role for R-chemotherapy plus radiation therapy?
Radiation therapy
Long-term disease control within radiation fields can be achieved in a significant number of patients with indolent stage I or stage II NHL by using dosages of radiation that usually range from 25 Gy to 40 Gy to involved sites or to extended fields that cover adjacent nodal sites.
Almost half of all patients treated with radiation therapy alone will relapse out-of-field within 10 years.
Rituximab with or without chemotherapy
For symptomatic patients who require therapy, when radiation therapy is contraindicated or when an alternative treatment is preferred, rituximab with or without chemotherapy can be employed (as outlined below for more advanced-stage patients).
The value of adjuvant treatment with radiation to decrease relapse, plus rituximab either alone or in combination with chemotherapy, has been extrapolated from trials of patients with advanced-stage disease and has not been confirmed.
Watchful waiting
Watchful waiting can be considered for asymptomatic patients.
Watchful waiting has never been compared with upfront radiation therapy in a prospective randomized trial;
a retrospective analysis of the Surveillance, Epidemiology and End Results Program (SEER) database over 30 years showed improved outcomes for upfront radiation therapy.
Other therapies as designated for patients with advanced-stage disease
Patients with involvement that is not able to be encompassed by radiation therapy are treated as outlined for patients with stage III or stage IV low-grade lymphoma.
Treatment for Indolent, Noncontiguous Stage II/III/IV Adult NHL
Optimal treatment of advanced stages of low-grade non-Hodgkin lymphoma (NHL) is controversial because of low cure rates with the current therapeutic options.
Numerous clinical trials are in progress to settle treatment issues, and patients are urged to participate. The rate of relapse is fairly constant over time, even in patients who have achieved complete response to treatment. Indeed, relapse may occur many years after treatment.
Currently, no randomized trials provide guidance to clinicians about the initial choice of watchful waiting, rituximab, nucleoside analogs, alkylating agents, combination chemotherapy, radiolabeled monoclonal antibodies, or combinations of these options. [Level of evidence: 1iiDiii]
For patients with indolent, noncontiguous stage II and stage III NHL, central lymphatic radiation therapy has been proposed but is not usually recommended as a form of treatment.
Standard Treatment Options for Indolent, Noncontiguous Stage II/III/IV Adult NHL
1. Watchful waiting for asymptomatic patients.
2. Rituximab.
3. Idelalisib.
4. Purine nucleoside analogs.
5. Alkylating agents (with or without steroids).
6. Bendamustine.
7. Combination chemotherapy.
8. Yttrium Y 90-ibritumomab tiuxetan.
9. Maintenance rituximab.
Watchful waiting for asymptomatic patients
The rate of relapse is fairly constant over time, even in patients who have achieved complete responses to treatment. Indeed, relapse may occur many years after treatment.
In this category, deferred treatment (i.e., watchful waiting until the patient becomes symptomatic before initiating treatment) should be considered.
The Follicular Lymphoma International Prognostic Index (FLIPI) and the revised FLIPI-2 can predict progression-free survival (PFS) and overall survival (OS), but the scores cannot be used to establish the need for therapy in asymptomatic patients.
Evidence (watchful waiting):
1. Three randomized trials compared watchful waiting with immediate chemotherapy.[Level of evidence: 1iiA]
• All three trials showed no difference in cause-specific or OS.
• For patients randomly assigned to watchful waiting, the median time to require therapy was 2 to 3 years and one-third of patients receiving watchful waiting never required treatment with watchful waiting (half died of other causes and half remained progression free after 10 years).
2. A selected group of 107 patients with advanced-stage follicular lymphoma were managed with initial watchful waiting; with a median delay of 55 months, subsequent therapy resulted in equivalent freedom from treatment failure and OS compared with a similar cohort treated immediately with rituximab.[Level of evidence: 3iiiDiii] This implies that watchful waiting remains a relevant approach even in the rituximab era.
Rituximab
Rituximab may be considered as first-line therapy, either alone or in combination with other agents.
• Rituximab alone, as was shown in the ECOG-E4402 (NCT00075946) trial, for example.
• R-bendamustine: rituximab + bendamustine.
• R-F: rituximab + fludarabine.
• R-CVP: rituximab + cyclophosphamide + vincristine + prednisone.
• R-CHOP: rituximab + cyclophosphamide + doxorubicin + vincristine + prednisone. A Cochrane meta-analysis could not identify any OS benefit of adding doxorubicin to chemotherapy regimens with rituximab or to chemotherapy regimens without rituximab.[Level of evidence: 1iiA]
• R-FM: rituximab + fludarabine + mitoxantrone.
• R-FCM: rituximab + fludarabine + cyclophosphamide + mitoxantrone.
Standard therapy includes rituximab, either alone or in combination with purine nucleoside analogs such as fludarabine or 2-chlorodeoxyadenosine, alkylating agents (with or without steroids), or combination chemotherapy.
A prospective, randomized trial of 534 patients with previously untreated, advanced-stage follicular lymphoma compared R-CHOP, R-FM, and R-CVP. With a median follow-up of 34 months, there was no difference in OS, but the 3-year PFS favored R-CHOP (68%) and R-FM (63%) over R-CVP (52%) (P for the three regimens = .011).[Level of evidence: 1iiDiii]
Evidence (rituximab):
• Four randomized, prospective studies of previously untreated patients (involving more than 1,300 patients) and one Cochrane meta-analysis including both untreated and previously treated patients (involving almost 1,000 patients) have compared rituximab plus combination chemotherapy with chemotherapy alone.[Level of evidence: 1iiA]
• Rituximab plus chemotherapy was superior in terms of event-free survival (EFS) or PFS (ranging from 2–3 years) in all of the studies and in terms of OS in all but one study (absolute benefit ranging from 6%–13% at 4 years, P < .04 and hazard ratio [HR] = 0.63 [0.51–0.79] for the meta-analysis).
• All of these trials were performed in symptomatic patients who required therapy. These results do not negate watchful waiting when appropriate.
• 18F-FDG PET-CT (fluorine F 18-fludeoxyglucose positron emission tomography–computed tomography) scan status at the completion of rituximab plus chemotherapy induction therapy is strongly predictive of outcome. It is not yet known whether acting on the results of the scans translates into better outcomes.
Idelalisib
Idelalisib is a selective inhibitor of PI3kδ, an integral part of the B-cell receptor.
Among 64 patients with heavily pretreated relapsed indolent NHL, idelalisib achieved an overall response of 47%, with a median duration of response rate of 18 months.[Level of evidence: 3iiiDiv]
Purine nucleoside analogs
• Fludarabine.
• 2-chlorodeoxyadenosine.
Alkylating agents (with or without steroids)
• Cyclophosphamide (oral or intravenous).
• Chlorambucil (oral).
Bendamustine
Evidence (bendamustine):
1. In a prospective randomized trial NCT00991211, 527 patients with indolent and mantle cell lymphoma were randomly assigned to a bendamustine-plus-rituximab arm versus an R-CHOP arm.[Level of evidence: 1iiDiii]
• With a median follow-up of 45 months, the median PFS favored the bendamustine arm (69 months vs. 31 months [HR, 0.58; 95 % confidence intervaI (CI), 0.44–0.74; P < .0001]) but with no difference in OS.
• The bendamustine arm was associated with significantly lower rates of alopecia, hematologic toxicity, stomatitis, peripheral neuropathy, and infections than was the R-CHOP arm.
Combination chemotherapy
• CVP: cyclophosphamide + vincristine + prednisone.
• CVP followed by rituximab maintenance.
• C-MOPP: cyclophosphamide + vincristine + procarbazine + prednisone.
• CHOP: cyclophosphamide + doxorubicin + vincristine + prednisone. A Cochrane meta-analysis could not identify any OS benefit to adding doxorubicin to chemotherapy regimens with or without rituximab.[Level of evidence: 1iiA]
• FND: fludarabine + mitoxantrone +/- dexamethasone, as evidenced in the SWOG-9501 (NCT00002670) trial, for example.
Yttrium Y 90-ibritumomab tiuxetan
Yttrium Y 90 (90Y)-ibritumomab tiuxetan is available for previously untreated and relapsing patients with minimal (<25%) or no marrow involvement with lymphoma (iodine I 131 [131I]-tositumomab is no longer available because of commercial disengagement).
In a randomized, prospective trial, 554 patients with previously untreated advanced-stage follicular lymphoma received either R-CHOP times six cycles or CHOP times six cycles followed by 131I-tositumomab radioimmunotherapy (RIT); with a median follow-up of 4.9 years, there was no significant difference between the PFS and OS (2-year OS, R-CHOP, 97%; CHOP-RIT, 93%; P = .08).[Level of evidence: 1iiD] Because a significant prolongation of PFS was seen for R-CHOP followed by rituximab maintenance compared with R-CHOP alone, this lack of benefit for CHOP-RIT was particularly disappointing. 131I-tositumomab became commercially unavailable in 2013.
In a randomized trial of 409 patients with stage III or IV follicular lymphoma who achieved a complete or partial response, 90Y-ibritumomab tiuxetan consolidation versus no consolidation was evaluated. The radiolabeled antibody consolidation improved median PFS by 3 years (P < .001), and median time to next treatment was improved by 5.1 years (P < .001); however, there was no change in OS.[Level of evidence: 1iiDiii]
Maintenance rituximab
After induction therapy with rituximab only or with rituximab plus chemotherapy, rituximab can be used once every 2 to 3 months. Several studies have evaluated this approach.
Evidence (maintenance rituximab for previously untreated patients):
1. In the PRIMA (NCT00140582) study, 1,019 high-risk, previously untreated, symptomatic patients achieved complete or partial response after induction therapy with immunochemotherapy (usually R-CHOP) and were then randomly assigned to 2 years of maintenance rituximab versus no maintenance.[Level of evidence: 1iiDiii]
• With a median follow-up of 36 months, PFS favored rituximab maintenance 74.9% to 57.6% (HR, 0.56; 95% CI, 0.44–0.68; P < .0001) but with no difference in OS.
2. In the United Kingdom/International study (NCT00112931), 379 previously untreated patients with asymptomatic, low-burden disease were randomly assigned to watchful waiting versus rituximab induction only versus rituximab induction followed by 2 years of rituximab maintenance.[Level of evidence: 1iiC]
• Although OS and histologic transformation rates were no different at 3 years, maintenance rituximab was favored based on quality-of-life studies (Mental Adjustment to Cancer Scale P = .0004 at 7 months; Illness Coping Score P = .0012 at 7 months) and time-to-initiation of new treatment by 3 years (54% for watchful waiting vs. 12% for rituximab maintenance [HR, 0.21; 95% CI, 0.14–0.31; P < .0001]).[Level of evidence: 1iiC]
• This study suggested that for some patients, watch and wait resulted in watch and worry. However, from the perspective of OS and histologic transformation rates, no benefit could be seen with rituximab maintenance.
3. In the RESORT (NCT00075946) study, 289 previously untreated patients with asymptomatic, low-burden disease were randomly assigned to rituximab induction alone, with a re-treatment strategy that used rituximab at relapse, or rituximab induction plus maintenance rituximab every 13 weeks until treatment failure.[Level of evidence: 1iiC]
• With a median follow-up of 4.5 years, there was no difference in median time-to-treatment failure (defined as failing rituximab alone) or in health-related quality of life. A re-treatment strategy achieved comparable disease control using significantly fewer doses of rituximab.
These three randomized trials in previously untreated patients show no advantage for the use of rituximab maintenance versus observation and reinduction of therapy at the time of relapse. The trials suggest a benefit for maintenance rituximab after reinduction for relapsed disease.
Many questions remain about rituximab maintenance, particularly about truncating therapy at 2 years and long-term safety and efficacy.
A trial extending rituximab maintenance to 5 years showed similar EFS or OS versus 1 year of maintenance after induction therapy with rituximab in previously untreated patients.[Level of evidence: 1iiA]
4. In a trial that studied the use of induction without rituximab (cyclophosphamide, vincristine, prednisone), 387 patients were randomly assigned to receive 2 years of rituximab maintenance.[Level of evidence: 1iiDiii]
• With a median follow-up of 11.5 years, there was improved PFS with maintenance (4.8 years vs. 1.3 years; HR, 0.49; P < .0001), but no difference in OS (10-year OS, 59%–67%).
For previously untreated patients, all of the studies showed improvement of PFS, with no change in OS.
Evidence (maintenance rituximab for previously treated patients):
1. In a prospective, randomized trial of 465 patients with relapsed follicular lymphoma, responders to R-CHOP or CHOP were further randomly assigned to receive rituximab maintenance (one dose every 3 months for 2 years) or no maintenance.[Level of evidence: 1iiDiii]
• At 6 years' median follow-up, rituximab maintenance was better for median PFS (44 months vs. 16 months, P < .001) and borderline for 5-year OS (74% vs. 64%, P = .07).
• This benefit for maintenance was evident even for patients who received rituximab during induction therapy. Most patients in both arms received extensive rituximab during postprotocol salvage treatment.
2. In a prospective, randomized trial of 280 patients with relapsed follicular lymphoma, responders to chemotherapy and autologous stem cell transplantation consolidation were randomly assigned to receive four doses of rituximab maintenance or no maintenance.[Level of evidence: 1iiDiii]
• With an 8.3-year median follow-up, the 10-year PFS favored maintenance (54% vs. 37% [HR, 0.66; 95% CI, 0.47–0.91; P = .012]), but there was no difference in OS.
3. A meta-analysis of nine randomized clinical trials with a total of 2,586 patients with follicular lymphoma, most of whom had relapsed disease, compared rituximab maintenance with no maintenance and showed improved OS for rituximab maintenance in previously treated patients (HRdeath, 0.72; 95% CI, 0.57–0.91).[Level of evidence: 1iiA]
For previously treated patients, there is more evidence to suggest an OS advantage with the use of rituximab maintenance.
Treatment Options Under Clinical Evaluation for Indolent, Noncontiguous Stage II/III/IV Adult NHL
Because none of the standard therapies listed above are curative for advanced-stage disease, innovative approaches are under clinical evaluation.
The approaches include intensive therapy with chemotherapy and total-body irradiation (TBI) followed by autologous or allogeneic bone marrow transplantation (BMT) or peripheral stem cell transplantation (PSCT), and the use of idiotype vaccines and radiolabeled monoclonal antibodies.
1. Intensive therapy with chemotherapy with or without TBI or high-dose radioimmunotherapy followed by autologous or allogeneic BMT or PSCT is under clinical evaluation.
2. Phase III trials comparing chemotherapy alone versus chemotherapy followed by anti-idiotype vaccine.
3. Extended-field radiation therapy (stage III patients only).
4. Ofatumumab—human anti-CD20 monoclonal antibody.
5. Short-course low-dose, palliative radiation therapy (2 × 2 Gy).
Treatment for Indolent, Recurrent Adult NHL
In general, treatment with standard agents rarely produces a cure in patients whose disease has relapsed. Sustained remissions after relapse can often be obtained in patients with indolent lymphomas, but relapse will usually ensue.
Favorable survival after relapse has been associated with an age younger than 60 years, complete remission rather than partial remission, and duration of response longer than 2 years.
Even the most favorable subset, however, has a tenfold greater mortality compared with age-adjusted U.S. population rates.
Patients who experience a relapse with indolent lymphoma can often have their disease controlled with single agent or combination chemotherapy, rituximab (an anti-CD20 monoclonal antibody), lenalidomide, radiolabeled anti-CD20 monoclonal antibodies, or palliative radiation therapy.
Long-term freedom from second relapse, however, is uncommon and multiple relapses will usually occur.
Patients with indolent lymphoma may experience a relapse with a more aggressive histology. If the clinical pattern of relapse suggests that the disease is behaving in a more aggressive manner, a biopsy should be performed. Documentation of conversion to a more aggressive histology requires an appropriate change to therapy applicable to that histologic type. Rapid growth or discordant growth between various disease sites may indicate a histologic conversion.
In a retrospective review of 325 patients between 1972 and 1999, the risk of histologic transformation was 30% by 10 years from diagnosis. In this series, high risk factors for subsequent histologic transformation were advanced stage, high-risk Follicular Lymphoma International Prognostic Index, and expectant management. The median survival after transformation was 1 to 2 years, with 25% of patients alive at 5 years and with approximately 10% to 20% of patients alive 10 years after re-treatment.
A prospective trial of 631 patients with follicular lymphoma and with a median follow-up of 60 months in the rituximab era (2002–2009) found a 5-year transformation rate (11%) to a higher-grade histology. The median overall survival (OS) after transformation was 50 months, and the 5-year OS rate was 66%, if the transformation occurred more than 18 months after a diagnosis of follicular lymphoma. This series describes a better prognosis for patients with transformation than was experienced by patients in the pre-rituximab era.
(Refer to the Treatment for Aggressive, Recurrent Adult NHL section of this summary for descriptions of the regimens used to treat histologic conversions.) The durability of the second remission may be short, and clinical trials should be considered.
Standard Treatment Options for Indolent, Recurrent Adult NHL
1. Chemotherapy (single agent or combination).
2. Rituximab.
3. Obinutuzumab.
4. Lenalidomide.
5. Radiolabeled anti-CD20 monoclonal antibodies.
6. Palliative radiation therapy.
Chemotherapy (single agent or combination)
Significant activity for fludarabine and 2-chlorodeoxyadenosine has been demonstrated in relapsed low-grade lymphomas, both as single agents and in combination with other drugs.
Patients may respond to the original induction regimen again, especially if the duration of remission exceeds 1 year.
For relapsing patients, rituximab alone or in combination with agents not previously used may induce remissions.
Rituximab
Rituximab results in a 40% to 50% response rate in patients who relapse with indolent B-cell lymphomas. Rituximab can also be combined with combination chemotherapy.
Evidence (rituximab):
• In three randomized, prospective studies involving previously treated patients with relapsed indolent lymphoma, patients were randomly assigned to rituximab maintenance after re-treatment with combination chemotherapy (with or without rituximab during induction) or rituximab alone; all trials showed prolongation of response duration, and one trial demonstrated improvement in median progression-free survival (PFS) (3.7 years vs. 1.3 years, P < .001) and OS (74% vs. 64%, P = .07) at 5 years with a median follow-up of 39 months favoring maintenance rituximab.
Obinutuzumab
Obinutuzumab is a CD20-binding monoclonal antibody with alternative epitope binding.
Evidence (obinutuzumab):
1. In a randomized prospective trial involving 396 patients with rituximab-refractory indolent lymphoma (mostly follicular lymphoma), patients received obinutuzumab plus bendamustine followed by obinutuzumab maintenance therapy versus bendamustine alone with no maintenance therapy.[Level of evidence: 1iiDiii]
• With a median follow-up of 21.9 months, the PFS was significantly longer in the obinutuzumab group, demonstrating a median PFS not reached (95% CI, 22.5 months to not reached) versus 14.9 months for the control arm of bendamustine (95% CI, 12.8‒16.6 months).
• There was no difference noted in OS.
Lenalidomide
Responses of 20% to 56% have been reported for lenalidomide, especially in patients with follicular lymphoma and small lymphocytic lymphoma, with even higher responses noted for the combination of lenalidomide and rituximab.[Level of evidence: 3iiiDiv]
Radiolabeled anti-CD20 monoclonal antibodies
Durable responses to radiolabeled monoclonal antibodies, such as yttrium Y 90 (90Y)-ibritumomab tiuxetan (commercially available) and iodine I 131-tositumomab (commercially unavailable), have also been reported before and after cytotoxic chemotherapy.[Level of evidence: 1iiDiii]
A prospective trial of 409 patients with follicular lymphoma who responded to induction chemotherapy were randomly assigned to 90Y-ibritumomab tiuxetan or no further consolidation; with a median follow-up of 7.3 years, the 8-year PFS favored 90Y-ibritumomab tiuxetan (41% vs. 22% [HR, 0.47; P < .001), but there was no difference in OS.[Level of evidence: 1iiDiii]
Palliative radiation therapy
Palliation may be achieved with very low-dose (4 Gy) involved-field radiation therapy in two fractions for patients with indolent and aggressive relapsed disease. In a prospective randomized trial, treatment with 4 Gy was inferior to treatment with 24 Gy in 12 fractions in PFS (77% vs. 92%, P < .0001).[Level of evidence: 1iiDiii]
Treatment Options Under Clinical Evaluation for Indolent, Recurrent Adult NHL
• Stem cell transplant. In many institutions, autologous or allogeneic stem cell transplantations (ASCT) are being used for patients whose disease has relapsed. Such an approach is still under evaluation but should be considered in the context of a clinical trial.
Evidence (stem cell transplant):
• The German Low-Grade Lymphoma Study Group treated 307 patients with follicular lymphoma with two cycles of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone)-like induction chemotherapy and then randomly assigned them to ASCT versus interferon maintenance. With a median follow-up of 4.2 years, the 5-year PFS was 65% for transplantation versus 33% for interferon (P < .001), but with no difference in OS.[Level of evidence: 1iiDiii]
Treatment for Aggressive, Stage I and Contiguous Stage II Adult NHL
Patients with stage I or contiguous stage II diffuse large B-cell lymphoma are candidates for combination chemotherapy with or without involved-field radiation therapy (IF-XRT).
Recommendation: R-CHOP given every 2 weeks for six cycles, with or without IF-XRT.
Standard Treatment Options for Aggressive, Stage I and Contiguous Stage II Adult NHL
R-CHOP with or without IF-XRT.
Four prospective randomized trials have evaluated the comparison of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) or more intensive CHOP-based chemotherapy alone versus combined–modality therapy with CHOP and IF-XRT.
Evidence (CHOP vs. CHOP with IF-XRT):
1. In a randomized trial with 7 years' median follow-up, 576 patients older than 60 years with early-stage disease received four cycles of CHOP with or without IF-XRT; there was no difference in 5-year event-free survival (EFS) (61% vs. 64%, P = .5) or overall survival (OS) (72% vs. 68%, P = .6).[Level of evidence: 1iiA]
2. A randomized trial of 401 patients comparing eight cycles of CHOP with three cycles of CHOP with IF-XRT was initially reported as having an OS advantage for the combined-modality arm at 5 years, but a reevaluation for OS at 18 years showed no difference in either arm of the study.[Level of evidence: 1iiA]
3. A randomized study (EST-1484) of 210 patients who attained a radiologic complete remission after eight cycles of CHOP compared IF-XRT with no further therapy; there was no difference in OS at 10 years (68% vs. 65%, P = .24).[Level of evidence: 1iiA]
4. A randomized trial of 631 patients younger than 60 years compared more intensive CHOP-based chemotherapy versus three cycles of CHOP with IF-XRT; with 4 years' median follow-up, the intensive chemotherapy was superior in 5-year EFS (82% vs. 74%, P > .001) and 5-year OS (90% vs. 81%, P = .001).[Level of evidence: 1iiA]
The confirmation of efficacy for rituximab in advanced-stage disease as evidenced in SWOG-S0014 (NCT00005089), for example, has suggested the use of R-CHOP with or without radiation therapy but its use is only supported by retrospective comparisons.[Level of evidence: 3iiiDiii]
• R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone); four to six cycles.
• R-CHOP (three to six cycles) + IF-XRT.
Evidence (R-CHOP vs. CHOP):
• A randomized study (DSHNHL-1999-1A [NCT00052936]) of 1,222 patients older than 60 years compared R-CHOP given every 2 weeks for six or eight cycles with CHOP given every 2 weeks for six or eight cycles. Although patients with early-stage disease were included in this trial, most patients had advanced-stage disease.
• With a median follow-up of 72 months, the EFS favored R-CHOP given every 2 weeks for six or eight cycles (EFS at 6 y, 74% vs. 56%; P < .001).
• The OS favored R-CHOP for only six cycles because of increased toxicity in the eight-cycle arm (OS at 6 y, 90% vs. 80%, P = .0004).[Level of evidence: 1iiA]
• There has been no comparison to standard R-CHOP or CHOP given every 3 weeks. There are no comparative studies to establish an optimal number of chemotherapy cycles for patients with early-stage disease.
Treatment Options Under Clinical Evaluation for Aggressive, Stage I and Contiguous Stage II Adult NHL
• R-ACVBP (rituximab, doxorubicin, cyclophosphamide, vindesine, bleomycin, prednisone).
Treatment for Aggressive, Noncontiguous Stage II/III/IV Adult NHL
The treatment of choice for patients with advanced stages of aggressive non-Hodgkin lymphoma (NHL) is combination chemotherapy, either alone or supplemented by local-field radiation therapy.
The following drug combinations are referred to in this section:
• ACVBP: doxorubicin + cyclophosphamide + vindesine + bleomycin + prednisone.
• CHOP: cyclophosphamide + doxorubicin + vincristine + prednisone.
• CNOP: cyclophosphamide + mitoxantrone + vincristine + prednisone.
• m-BACOD: methotrexate + bleomycin + doxorubicin + cyclophosphamide + vincristine + dexamethasone + leucovorin.
• MACOP-B: methotrexate + doxorubicin + cyclophosphamide + vincristine + prednisone fixed dose + bleomycin + leucovorin.
• ProMACE CytaBOM: prednisone + doxorubicin + cyclophosphamide + etoposide + cytarabine + bleomycin + vincristine + methotrexate + leucovorin.
• R-CHOP: rituximab, an anti-CD20 monoclonal antibody, + cyclophosphamide + doxorubicin + vincristine + prednisone.
Standard Treatment Options for Aggressive, Noncontiguous Stage II/III/IV Adult NHL
1. R-CHOP.
2. Other combination chemotherapy.
R-CHOP
The following studies established R-CHOP as the standard regimen for newly diagnosed patients with diffuse large B-cell lymphoma (DLBCL). Dose intensification of R-CHOP by a 14-day versus a 21-day cycle did not result in improved outcomes.
Evidence (R-CHOP):
1. R-CHOP showed improvement in event-free survival (EFS) and overall survival (OS) compared with CHOP alone in 399 advanced-stage patients with DLBCL older than 60 years (EFS, 57% vs. 38%; P = .002, and OS, 70% vs. 57%; P = .007 at 2 years).[Level of evidence: 1iiA] At 10-years' median follow-up, the OS of patients who received R-CHOP compared with patients who received CHOP was 44% versus 28%, P < .0001.
2. Similarly, for 326 evaluable patients younger than 61 years, R-CHOP showed improvement in EFS and OS compared with CHOP alone (EFS, 79% vs. 59%, P = .001, and OS, 93% vs. 84%, P = .001 at 3 years).[Level of evidence: 1iiA]
3. A randomized study (DSHNHL-1999-1A [NCT00052936]) of 1,222 patients older than 60 years compared R-CHOP given every 2 weeks for six or eight cycles with CHOP given every 2 weeks for six or eight cycles. With a median follow-up of 72 months, the EFS favored R-CHOP given every 2 weeks for six or eight cycles (EFS at 6 years, 74% vs. 56%; P < .0001). The OS favored R-CHOP for only six cycles because of increased toxicity in the eight-cycle arm (OS at 6 years, 90% vs. 80%; P = .0004).[Level of evidence: 1iiA] There was no comparison with standard R-CHOP or CHOP given every 3 weeks.
4. A trial (NCT00140595) of 380 patients younger than 60 years with DLBCL and an age-adjusted International Prognostic Index (IPI) rating of 1 randomly assigned treatment of patients to ACVBP and rituximab (R-ACVBP) plus consolidation with methotrexate, ifosfamide, etoposide, and cytarabine versus CHOP and rituximab. With a median follow-up of 44 months, 3-year OS favored R-ACVBP (92% vs. 84%; hazard ratio, 0.44; 95% confidence interval (CI), 0.28–0.81, P = .007).[Level of evidence: 1iiA] The significantly worse toxicities with R-ACVBP, the narrow target population (<60 years with either elevated lactate dehydrogenase (LDH) or stage III-stage IV disease, but not both), and the lack of a confirmatory trial may inhibit adoption of R-ACVBP as a new standard of care.
Clinical trials continue to explore modifications of CHOP and rituximab with CHOP by increasing doses, reducing intervals between cycles, combining new drugs with new mechanisms of action, or applying extra doses of rituximab at the start and after completion of chemotherapy. None of these trials establishes a survival advantage for reduced intervals between cycles or for increasing doses of the chemotherapy.
Additive radiation therapy
After R-CHOP chemotherapy, involved-field radiation therapy has been proposed for initial bulky sites of disease (≥7.5 cm) or to extralymphatic sites; a nonrandomized cohort study (RICOVER-noRTH) suggested improved EFS, but not progression-free survival or OS.[Level of evidence: 3iiiDi] Future trials will focus on whether mid-treatment or end-of-treatment positron emission tomography–computed tomography can define whether radiation therapy should be used after systemic therapy.
Other combination chemotherapy
Doxorubicin-based combination chemotherapy produces long-term disease-free survival in 35% to 45% of patients. Higher cure rates have been reported in single-institution studies than in cooperative group trials.
Evidence (other combination chemotherapy):
1. A prospective, randomized trial of four regimens (CHOP, ProMACE CytaBOM, m-BACOD, and MACOP-B) for patients with DLBCL showed no difference in OS or time-to-treatment failure at 3 years.[Level of evidence: 1iiA]
2. Other randomized trials have confirmed no advantage among the standard doxorubicin-based combinations versus CHOP.[Level of evidence: 1iiA]
A randomized clinical trial failed to demonstrate a beneficial effect of adjuvant radiation therapy in advanced-stage aggressive NHL.
Stage IE or IIE gastric DLBCL
Four case series involving more than 100 patients with stage IE or IIE disease (with or without associated mucosa-associated lymphatic tissue) and with positive Helicobacter pylori infection reported that more than 50% of patients attained a durable complete remission after appropriate antibiotic therapy to eradicate H. pylori.[Level of evidence: 3iiiDiv]
Prognostic factors
An IPI for aggressive NHL (diffuse large cell lymphoma) identifies five significant risk factors prognostic of OS:
1. Age (≤60 years vs. >60 years).
2. Serum LDH (normal vs. elevated).
3. Performance status (0 or 1 vs. 2–4).
4. Stage (stage I or stage II vs. stage III or stage IV).
5. Extranodal site involvement (0 or 1 vs. 2–4).
Patients with two or more risk factors have a less than 50% chance of relapse-free survival and OS at 5 years. This study also identifies patients at high risk of relapse based on specific sites of involvement, including bone marrow, central nervous system (CNS), liver, lung, and spleen.
The bcl-2 gene and rearrangement of the myc gene or dual overexpression of the myc gene, or both, confer a particularly poor prognosis.
Patients at high risk of relapse may be considered for clinical trials. Molecular profiles of gene expression using DNA microarrays may help to stratify patients in the future for therapies directed at specific targets and to better predict survival after standard chemotherapy.
Treatment of tumor lysis syndrome
Patients with bulky and extensive lymphadenopathy and elevations of serum uric acid and LDH are at increased risk of tumor lysis syndrome resulting in metabolic derangements such as hyperuricemia, hyperkalemia, hyperphosphatemia, hypocalcemia, and subsequent acute renal failure. Treatment options include: alkaline hydration, allopurinol, and rasburicase, a recombinant urate oxidase.
CNS prophylaxis
CNS prophylaxis (usually with four to six injections of methotrexate intrathecally) is recommended for patients with paranasal sinus or testicular involvement.
Some clinicians are employing high-dose intravenous methotrexate (usually four doses) as an alternative to intrathecal therapy because drug delivery is improved and patient morbidity is decreased. CNS prophylaxis for bone marrow involvement is controversial; some investigators recommend it, and others do not.
• A retrospective analysis of 605 patients with diffuse large cell lymphoma who did not receive prophylactic intrathecal therapy identified an elevated serum LDH and more than one extranodal site as independent risk factors for CNS recurrence. Patients with both risk factors have a 17% probability of CNS recurrence at 1 year after diagnosis (95% CI, 7%–28%) versus 2.8% (95% CI, 2.7%–2.9%) for the remaining patients.[Level of evidence: 3iiiDiii]
Patients with diffuse, small, noncleaved-cell/Burkitt's lymphoma or lymphoblastic lymphoma have a 20% to 30% lifetime risk of CNS involvement. CNS prophylaxis is recommended for these histologies.
Treatment Options Under Clinical Evaluation for Aggressive, Noncontiguous Stage II/III/IV Adult NHL
• Bone marrow transplant (BMT) or stem cell transplantation (SCT).
Several randomized, prospective trials evaluated the role of autologous BMT or SCT consolidation versus chemotherapy alone in patients in first remission with diffuse large cell lymphoma.[Level of evidence: 1iiA] Although some of these trials demonstrated significant increases in EFS (by 10% to 20%) among patients who received high-dose therapy, significant differences in OS could not be demonstrated prospectively in any of the series.
Retrospective analyses of high-intermediate (two risk factors) or high-risk (more than three risk factors) patients as defined by IPI suggest improved survival with BMT in two of the trials. These studies do not establish that high-dose consolidation is of value to patients with aggressive lymphoma who are truly at high risk of relapse, and they also demonstrate that EFS may be a poor surrogate for OS for these patients.
• Radiation therapy consolidation to sites of bulky disease.
After R-CHOP induction chemotherapy (or similar regimens), the addition of involved-field radiation therapy to sites of initial bulky disease (≥5–10 cm) or to extralymphatic sites remains controversial. Increased risks, such as long-term toxicities (e.g., second malignancies), must be considered.
Treatment for Adult Lymphoblastic Lymphoma (ALL)
Lymphoblastic lymphoma is a very aggressive form of non-Hodgkin lymphoma (NHL), which often occurs in young patients, but not exclusively.
Lymphoblastic lymphoma is commonly associated with large mediastinal masses and has a high predilection for disseminating to bone marrow and the central nervous system (CNS).
The treatment paradigms are based on trials for ALL since lymphoblastic lymphoma and ALL are considered different manifestations of the same biologic disease. (Refer to the PDQ summary on Adult Acute Lymphoblastic Leukemia Treatment for more information.)
Treatment is usually patterned after ALL. Intensive combination chemotherapy with CNS prophylaxis is the standard treatment of this aggressive histologic type of NHL.
Radiation therapy is sometimes given to areas of bulky tumor masses. Since these forms of NHL tend to progress quickly, combination chemotherapy is instituted rapidly once the diagnosis has been confirmed.
The most important aspects of the pretreatment staging workup include careful review of the following pathological specimens:
• Bone marrow aspirate.
• Biopsy specimen.
• Cerebrospinal fluid cytology.
• Lymphocyte marker.
Standard Treatment Options for ALL
1.Intensive therapy.
2.Radiation therapy.
(Refer to the PDQ summary on Adult Acute Lymphoblastic Leukemia Treatment for more information.)
Intensive therapy
Standard treatment is intensive combination chemotherapy with CNS prophylaxis.
Radiation therapy
Radiation therapy is sometimes given to areas of bulky tumor masses.
Treatment Options Under Clinical Evaluation for ALL
New treatment approaches are being developed by the national cooperative groups. Other approaches include the use of bone marrow transplantation for consolidation. (Refer to the PDQ summary on Adult Acute Lymphoblastic Leukemia Treatment for more information.)
Treatment for Diffuse, Small Noncleaved-Cell/Burkitt Lymphoma
Diffuse, small, noncleaved-cell/Burkitt lymphoma typically involves younger patients and represents the most common type of pediatric non-Hodgkin Lymphoma.
Standard Treatment Options for Diffuse, Small Noncleaved-Cell/Burkitt Lymphoma
1. Aggressive multidrug regimens.
2. Central nervous system (CNS) prophylaxis.
Aggressive multidrug regimens
Standard treatment for diffuse, small, noncleaved-cell/Burkitt lymphoma is usually with aggressive multidrug regimens similar to those used for the advanced-stage aggressive lymphomas (such as diffuse large cell). Adverse prognostic factors include bulky abdominal disease and high serum lactate dehydrogenase.
Evidence (aggressive multidrug regimens):
• Aggressive combination chemotherapy patterned after that used in childhood Burkitt lymphoma has been very successful for adult patients. More than 60% of advanced-stage patients were free of disease at 5 years.
• Rituximab has been incorporated into these aggressive combination chemotherapy regimens. A nonrandomized, single-arm, prospective, multicenter trial of 363 patients, aged 16 years to 85 years, showed a 5-year progression-free survival of 71% and a 5-year overall survival of 80%.[Level of evidence: 3iiiA]
CNS prophylaxis
Patients with diffuse, small, noncleaved-cell/Burkitt lymphoma have a 20% to 30% lifetime risk of CNS involvement. CNS prophylaxis with methotrexate is recommended for all patients, usually given as four to six intrathecal injections. (Refer to the PDQ summary on Adult Acute Lymphoblastic Leukemia Treatment for more information).
Evidence (CNS prophylaxis):
• In a series of 41 patients treated with systemic and intrathecal chemotherapy, 44% of those who presented with CNS disease and 13% of those who relapsed with CNS involvement became long-term disease-free survivors. CNS relapse patterns were similar whether or not patients received radiation therapy, but increased neurologic deficits were noted among those patients who received radiation therapy.
Treatment for Aggressive, Recurrent Adult NHL
Bone marrow transplantation (BMT) is the treatment of choice for patients whose lymphoma has relapsed.
Standard Treatment Options for Aggressive, Recurrent Adult NHL
1. Bone marrow or stem cell transplantation.
2. Re-treatment with standard agents.
3. Palliative radiation therapy.
Bone marrow or stem cell transplantation
Bone marrow transplantation (BMT) is the treatment of choice for patients whose lymphoma has relapsed.
Preliminary studies indicate that approximately 20% to 40% of patients will have a long-term disease-free status, but the precise percentage depends on patient selection and the specific treatment used.
Preparative drug regimens have varied; some investigators also use total-body irradiation. Similar success has been achieved using autologous marrow, with or without marrow purging, and allogeneic marrow.
Evidence (BMT):
1. In a prospective, randomized study, (EORTC-PARMA), 215 patients in first or second relapse of aggressive lymphoma, younger than 60 years, and with no bone marrow or central nervous system involvement, were given two cycles of intensive combination chemotherapy. The 109 patients who responded were randomly assigned to receive four more cycles of chemotherapy and involved-field radiation therapy (IF-XRT) versus autologous BMT followed by IF-XRT. With a 5-year median follow-up, the event-free survival (EFS) was significantly improved with transplantation (46% vs. 12%). Overall survival (OS) was also significantly better with transplantation (53% vs. 32%).[Level of evidence: 1iiA] Salvage BMT was unsuccessful for patients on the nontransplant arm whose disease relapsed.
In general, patients who responded to initial therapy and who responded to conventional therapy for relapse before the BMT have had the best results.
2. In a prospective trial, patients who relapsed late (>12 months after diagnosis) had better OS than patients who relapsed earlier (8-year survival was 29% vs. 13%, P = .001).[Level of evidence: 3iiiA]
Peripheral stem cell transplantation (SCT) has yielded results equivalent to standard autologous transplantation. Even patients who never experienced complete remission with conventional chemotherapy may have prolonged progression-free survival (31% at 5 years) after high-dose chemotherapy and hematopoietic SCT if they retain chemosensitivity to reinduction therapy.[Level of evidence: 3iiiDiii] Some patients who relapse after a previous autologous transplantation can have durable remissions after myeloablative or nonmyeloablative allogeneic SCT.[Level of evidence: 3iiiDiv]
Evidence (peripheral SCT):
• In a randomized prospective trial, 396 patients with diffuse large B-cell lymphoma in first relapse or who were refractory to first-line therapy received either R-ICE (rituximab, ifosfamide, etoposide, and carboplatin) or R-DHAP (rituximab, dexamethasone, high-dose cytarabine, and cisplatin) followed by autologous SCT; there was no difference in 3-year EFS or OS.[Level of evidence: 1iiA]
• In a randomized prospective trial, 619 patients with relapsed or refractory aggressive lymphoma received either R-DHAP or R-GDP (rituximab, gemcitabine, dexamethasone, and cisplatin) followed by autologous SCT; at a median follow-up of 53 months, there was no difference in EFS or OS, but patients who received R-GDP reported less toxicity.[Level of evidence: 1iiC]
Re-treatment with standard agents
In general, re-treatment with standard agents rarely produces a cure in patients whose lymphomas relapse. Several salvage chemotherapy regimens are available.
• Rituximab alone can induce responses in 33% of patients with relapsing aggressive lymphoma of appropriate phenotype (CD20-positive).[Level of evidence: 3iiiDiv]
• Radiolabeled anti-CD20 monoclonal antibodies, such as iodine I 131-tositumomab (no longer commercially available) and yttrium Y 90-ibritumomab tiuxetan, induce 60% to 80% response rates in patients with relapsed or refractory B-cell lymphoma.[Level of evidence: 3iiiDiv]
• In two phase II trials, 49 patients showed a 19% to 35% overall response rate to lenalidomide with or without rituximab.[Level of evidence: 3iiiDiv]
Durable responses to radiolabeled monoclonal antibodies have been reported for transformed low-grade B-cell lymphoma. Not infrequently, an aggressive lymphoma may relapse as a small cell (indolent) lymphoma. Such a situation occurs with indolent lymphoma in the bone marrow and aggressive lymphoma in a nodal site. Patients may present in such a manner, and chemotherapy might successfully eradicate the peripheral disease while failing to eliminate the small cell component from the bone marrow. The clinical significance and natural history of this pattern of disease is not well defined.
Palliative radiation therapy
In general, patients with aggressive lymphoma who relapse with indolent histology will benefit from palliative therapy. Palliation may be achieved with very low-dose (4 Gy) IF-XRT for patients with indolent and aggressive relapsed disease.
Treatment Options Under Clinical Evaluation for Aggressive, Recurrent Adult NHL
• SCT. The indolent lymphomas may relapse with an aggressive histology (i.e., histologic conversion). The durability of the second remission may be short, and clinical trials, such as autologous or allogeneic peripheral SCT, should be considered.
NHL During Pregnancy
Non-Hodgkin lymphomas (NHL) occur more frequently than Hodgkin lymphoma in an older population. This age difference may account for fewer reports of NHL in pregnant patients.
Stage Information for NHL During Pregnancy
To avoid exposure to ionizing radiation, magnetic resonance imaging is the preferred tool for staging evaluation.
| Stage | Standard Treatment Options |
| Indolent NHL During Pregnancy | Delay treatment until after delivery |
| Aggressive NHL During Pregnancy | Immediate therapy |
| Early delivery, when feasible | |
| Termination of pregnancy |
Indolent NHL During Pregnancy
Treatment may be delayed for those women with an indolent NHL.
Aggressive NHL During Pregnancy
1. Immediate therapy
According to anecdotal case series, most NHL in pregnant patients are aggressive, and delay of therapy until after delivery appears to have poor outcomes. Consequently, some investigators favor immediate therapy, even during pregnancy.
In a review of 121 patient case reports from 74 papers, one-half of the patients had very aggressive lymphomas, such as Burkitt lymphoma, and one-half of the patients had involvement of the breast, ovaries, uterus, or placenta. One-half of the patients received therapy antepartum, and the 6-month survival was reported at 53%, with a live-birth rate of 83%.[Level of evidence: 3iiiDiv]
A multicenter retrospective analysis of 50 patients described pregnancy termination in 3 patients, deferral of therapy to postpartum in 15 patients (median 30 weeks gestation), and antenatal therapy applied to the remaining 32 patients (median 21 weeks gestation, all done after the first trimester). With a median follow-up of 41 months, the 3-year progression-free survival was 53%, and overall survival was 82%, using R-CHOP (rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone) or modifications of this regimen.[Level of evidence: 3iiiDiv]
2. Early delivery when feasible
For some women, early delivery, when feasible, may minimize or avoid exposure to chemotherapy or radiation therapy.
3. Termination of pregnancy
Termination of pregnancy in the first trimester may be an option that allows immediate therapy for women with aggressive NHL.
Evidence (treatment effect on children exposed in utero):
• With follow-up ranging from several months to 11 years, children who were exposed to high-dose doxorubicin-containing combination chemotherapy in utero (especially during the second and third trimester) have been found to be normal. For most of the chemotherapeutic agents used for the treatment of NHL, there are no data regarding long-term effects on children exposed in utero.
Treatment Options for Follicular Lymphoma
Treatment Options for Follicular Lymphoma (Grade 1-2)
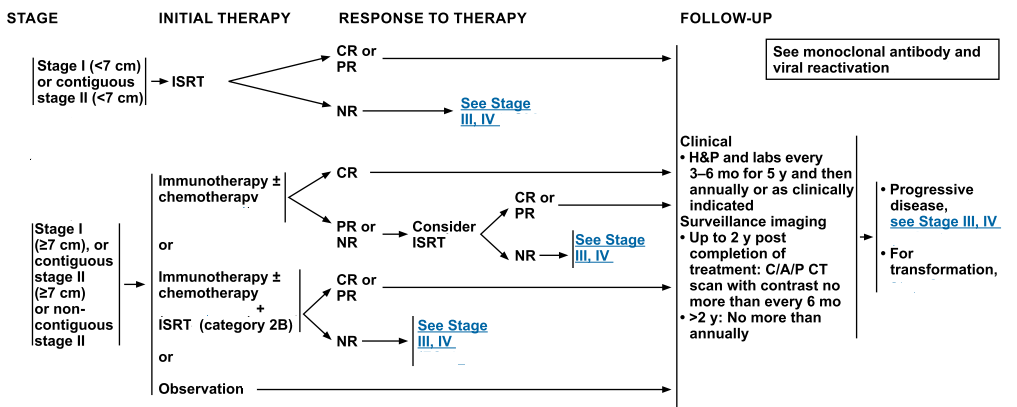

For Those without an Indication to Treat:
• Three large randomized trials comparing immediate treatment to delayed therapy in low burden, asymptomatic patients failed to show a survival benefit to early therapy.
• Median time to initiation of therapy over 3 decades remains 2.5 3.0 years in those being “watched.”
• Forty percent of patients over the age of 70 never require treatment.
• If treated with rituximab, indefinite maintenance was not better than retreatment (RESORT Trial) in asymptomatic patients. No QoL benefit.
Treatment of Early Stage FL
• Radiotherapy has been the recommended strategy for treatment of localized FL, based on data showing that 40‐50% of patients will have very long remissions –possibly cures – with this approach.
• Staging with PET is likely to better select patients for radiotherapy, improving outcomes.
• Involvedsite radiation therapy (ISRT) remains the recommended strategy for PET/CT Staged I FL.
• Alternatives, ranging from observation to immunotherapy +chemotherapy with or without ISRT, are associated with excellent outcomes. ISRT required for cure.
• Therapy should be individualized based on site of disease and patient age and preference.
Idications for Treatment for Advanced Stage FL: Stage III, IV
• Symptoms
• Threatened end‐organ function, ascites, effusions
• Cytopenia secondary to lymphoma
• Bulky disease (>7 cm) --- (3 masses > 3 cm )
• Steady progression --- (Rapid progression: >50% increase in 6 months)
• Patient preference (?)
• Candidate for clinical trial
Others:
• Splenomegaly
• Presentation with concurrent histologic transformation
End of Treatment Response
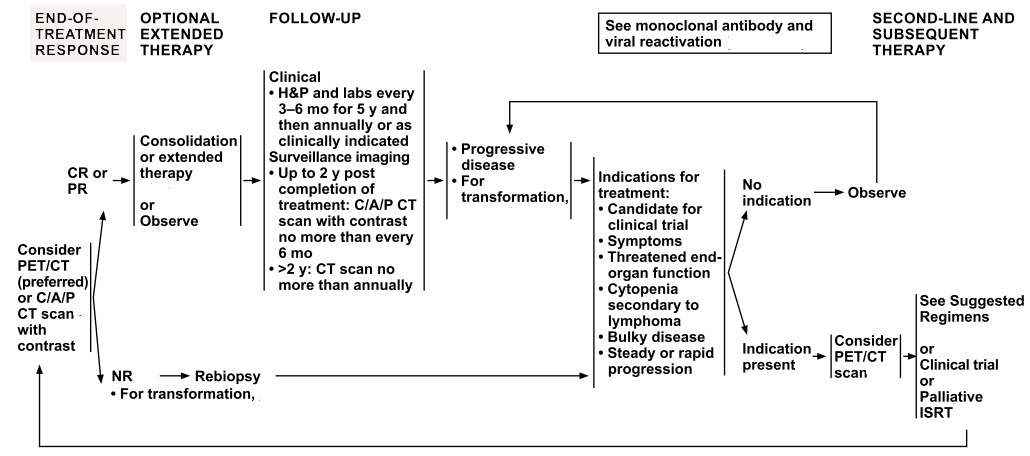
FL Patients who progress within 24 mo of R‐CHOP have a 5 yr OS of only 50%.
Based on the results of meta‐analysis and the supporting phase III trials, rituximab in combination with chemotherapy is the STANDARD OF CARE patient patient requiring therapy. (CATEGORY 1)
The optimal R‐CHEMO regimen remains undefined.
Best Induction therapy: R‐bendamustine?
Suggestive Treatment Regimens for Follicular Lymphoma (Grade 1-2)

B-R regimen (bendamustine + rituximab) is our first choice.
rituximab 375 mg/m2 IV on day 1
bendamustine 90 mg/m2 IV on days 1 and 2
(Repeated every 28 days for up to 6 cycles)

Treatment of Relapsed/Refractory FL: Novel Agents/strategies
• PI3K inhibitors
• Lenalidomide
• BTK inhibitors
• New antibodies/antibody‐drug conjugates(ADCs)
• BCL‐2 inhibitors
• Checkpoint inhibitors
• Hematopoietic cell transplant (HCT)
• CAR T‐cell therapy
DYNAMO Trial of Duvelisib in Double Refractory Indolent Lymphoma
Duvelisib is an dual inhibitor of P13K-delta and P13K-gamma
Percent change in nodal target lesions
88% of patients had reduction in target lymph nodes (IRC data)
Median Progression‐free survival (months): All iNHL (9.o), FL (8.3), SLL (11.7), MZL (14.3)
Copanlisib is a pan-class I PI3K inhibitor with activity against alpha and delta isoforms: CHRONOS‐1 trial In FL: ORR 58.7% CR 14.4% PR 44.2% Median PFS: 11.2 mo. Median DOR: 12.2
Sept 14, 2017: FDA granted accelerated approval for treatment of adults with relapsed FL who have failed at least 2 prior therapies
Pembrolizumab + Rituximab

Polatuzumab Vedotin* + Bendamustine + either Rituximab or Obinutuzumab (FL)

Transformation to Diffuse Large B-cell Lymphoma


Treatment Options for Diffuce Large B-Cell Lymphoma
First-line Therapy for Diffuce Large B-Cell Lymphoma
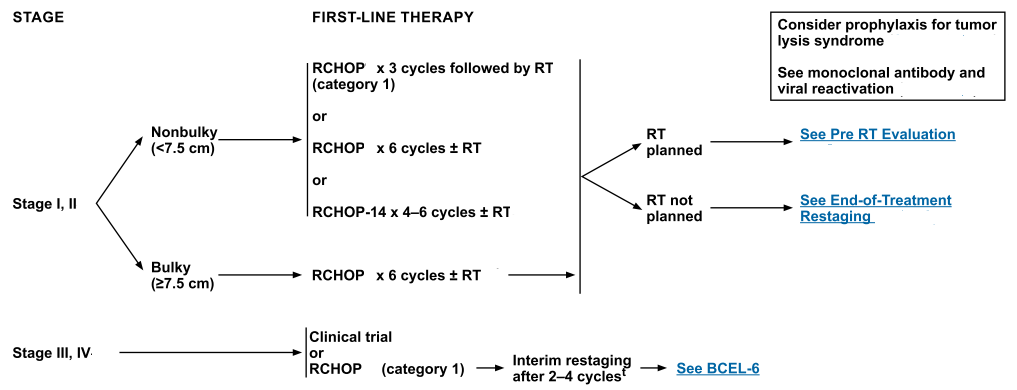
Pre RT Evaluation

End of Treatment Restaging

Interim Restaging

Relapse/Refractory Disease
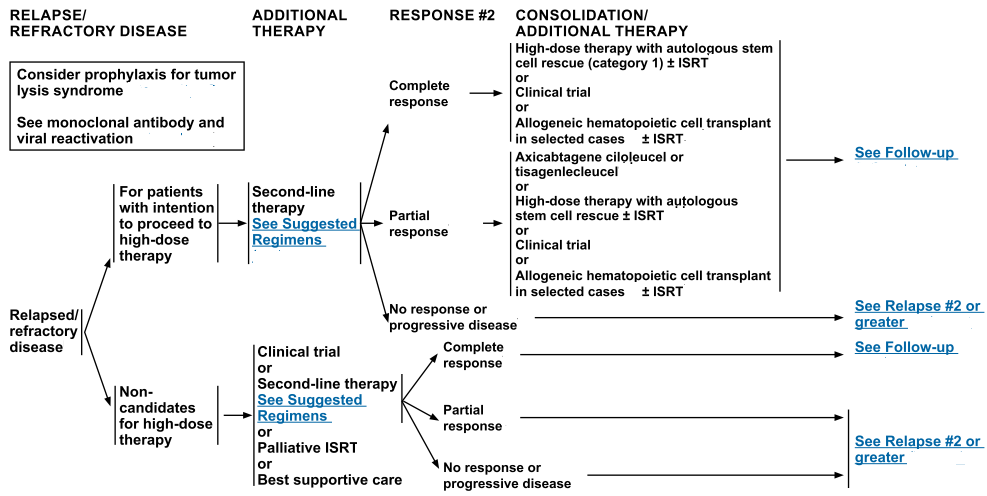
Follow-up
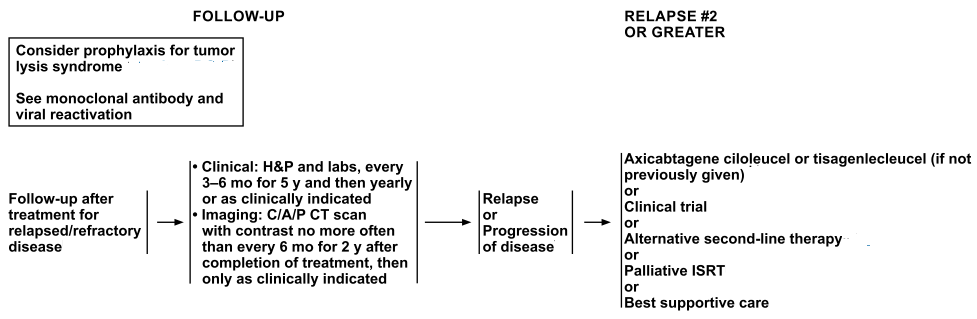
Prognostic Index

Prognostic Model to assess the risk of CNS Disease

Suggestive Treatment Regimens for Diffuce Large B-Cell Lymphoma

International Standard of Care: R‐CHOP
Rituximab 375 mg/m2 day 1
Cyclophosphamide 750 mg/m2 day 1
Doxorubicin 50 mg/m2 day 1
Vincristine 1.4 mg/m2 day 1 (2 mg max)
Prednisone 40 mg/m2 (or 100 mg) daily x 5
(Repeated every 21 days -- CHOP-21)
